- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
Progressive neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease are a major public health burden. Neurodegenerative diseases have a common pathophysiology of protein aggregate formation but the exact mechanism of neuronal destruction has not been pinpointed. Various theories have been proposed in the literature to explain the underlying causes of these diseases, including reactive oxygen species generation, mitochondrial dysfunction, excessive neuroinflammation, microglial cell dysfunction, and others. These neurodegenerative diseases appear to be multifactorial and complex in origin, and there are no treatments or approved drugs that can halt the disease progression. Recent genetic and basic science research into the mechanisms of neurodegenerative diseases have implicated a new paradigm that incorporates the previous theories into a more cohesive conceptual framework. A failure to balance neuronal autophagy and lysosomal function may be the common root cause of these disorders that can lead to mitochondrial dysfunction, excessive protein aggregation, increased reactive oxygen species generation and inflammation. This review examines the current research on autophagy and new drug and/or natural product approaches to enhancing partial autophagy that can help prevent neuronal death and the progression of neurodegenerative disease. The critical factor in brain health may be the balance of autophagy and cell death in maintaining viable neurons.
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
Proteostasis is the process of protein homeostasis within cells. Intracellular accumulations of proteins in neurons and other cells are maintained by protein degradation in proteasomes for short-lived proteins and in lysosomes for longer-lasting proteins.1 Recent research into the mechanisms of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease has demonstrated that a common pathway of neuronal cell death is misfolded protein accumulation.2 Intracellular accumulation of misfolded and aggregated proteins can trigger neuronal cell death and apoptosis. A number of factors contribute to protein misfolding, including increased damage to proteins by reactive oxygen species, mitochondrial dysfunction, decreased cellular antioxidant mechanisms (mediated by the Nrf2 pathway), increased inflammation and generation of the inflammasome, inhibition of ubiquitination and inhibition of proteasomal degradation. Interestingly, all of these mechanisms have been proposed as underlying or contributing causes in neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. One common hallmark of neurodegenerative diseases is the accumulation of insoluble protein aggregates inside and outside cells. In Parkinson’s disease, damaged proteins that have exposed hydrophobic residues (such as α-synuclein) can polymerize and lead to accumulations of intracellular protein aggregates. Long-lived cells such as neurons may be particularly sensitive to protein aggregates over time. A major mechanism for removing intracellular protein aggregates is autophagy, the process of the cell “eating” itself. Previously, it was thought that autophagy was a harmful process, triggered by low nutrient levels, that leads to cell death. However, new research has demonstrated that autophagy may be a critical process for cells to remove harmful protein aggregates and malfunctioning organelles (such as mitochondria). The removal of protein aggregates and malfunctioning mitochondria by lysosomal destruction may be essential for cell longevity. Low-dose rapamycin, low-calorie dietary restriction (nutrient deprivation), specific phytonutrients and other signals can stimulate autophagy, which helps neurons survive longer by enhancing lysosomal degradation of both misfolded protein aggregates and dysfunctional mitochondria.2 Intracellular and extracellular accumulations of α-synuclein (in both Parkinson’s disease and Alzheimer’s disease) and amyloid-β (Aβ) protein (along with neurofibrillary tangles of tau protein) are thought to be critical in the pathogenesis of Parkinson’s disease and Alzheimer’s disease.3
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
Alzheimer’s disease was estimated to affect 5.2 million Americans and close to 50 million worldwide in 2016, and it is the leading cause of dementia in the United States and the rest of the world.4,5 Alzheimer’s disease is also the sixth leading cause of death in the United States, and the disease burden and cost of care are expected to increase as the population ages. Alzheimer’s disease is the most common neurodegenerative disease; Parkinson’s disease is the second most common. The age-standardized prevalence of dementia ranges from 5% to 7% in most countries.6 Alzheimer’s disease is characterized by a progressive loss of mental function and memory as well as overall loss of neurons and actual brain mass. This loss of function is correlated with pathophysiological alterations, including the deposition of plaques (composed of Aβ protein aggregates and amyloidosis) and the formation of neurofibrillary tangles composed of hyperphosphorylated tau protein.7,8 Age is the primary risk factor for Alzheimer’s disease, and the risk doubles every 10 years after age 60, although the risk may be declining (slightly) in most industrialized countries.9 The risk for Alzheimer’s disease is greater for women over 60 years of age than for men, but the risk for Parkinson’s disease is greater for men. Genetic risk factors for Alzheimer’s disease include mutations in amyloid precursor protein (APP), presenelin-1, and presenilin-2 for early-onset disease, but these account for <1% of Alzheimer’s disease. Genetic risk factors for late-onset disease include apolipoprotein E (APOE). Carriers of one APOɛ4 allele are at 2- to 3-fold increased risk, and those with two ɛ4 alleles are at approximately 8- to 12-fold increased risk, compared with noncarriers.10 Other genes have been identified, but there is as yet no conclusive evidence of penetration or function in a significant percentage of patients with Alzheimer’s disease, so the disease is often referred to as multifactorial and environmentally modulated. There is a 10%–30% increased risk of late-onset Alzheimer’s disease in people with a first-degree relative with the disease,11 and this shows that most of the risk factors are environmental. A gene called triggering receptor expressed on myeloid cells 2 (TREM2) is associated with a rare autosomal recessive form of dementia.12 The TREM2 gene may be linked to the possible increased inflammation in the brain that is hypothesized as a possible underlying cause of Alzheimer’s disease because it can activate immune cells to secrete increased inflammatory cytokines such as TNF-α. Because Alzheimer’s disease has a long presymptomatic period, it may be possible for drugs, lifestyle changes such as diet and exercise, and phytonutrients to prevent or delay Alzheimer’s disease. Currently, there are no drugs that have been proven to prevent or delay dementia progression.
Because the genetic influence in late-onset Alzheimer’s disease is likely below 30%, many environmental risk factors have been studied and proposed as possible risk factors for the disease. Higher cholesterol levels have been correlated with an increased risk of Alzheimer’s disease,13 but statin drugs have not been shown to decrease that risk.14 Cerebrovascular disease often coexists with Alzheimer’s disease,15 and there is some correlation of atherosclerosis in the larger arteries with Alzheimer’s disease. Mechanistically, decreased blood flow to the brain could be a constant stressor for neurons, and increased blood flow to the brain is one of the proposed mechanisms of action of Ginkgo biloba.16 Both hypertension17 and obesity18 are strongly correlated with Alzheimer’s disease risk. Hypertension is thought to be a risk factor due primarily to cerebrovascular disease, whereas obesity is postulated to affect both inflammation and astrocyte function,19 in addition to atherosclerosis. Astrocytes are the most prevalent cells in the central nervous system (CNS) and act as support cells to help maintain neuronal function. Insulin resistance and type 2 diabetes also are major risk factors for Alzheimer’s disease.20 Lack of physical activity has also been shown to be a risk factor. A meta-analysis showed a 45% reduction in Alzheimer’s disease among those who were physically active compared with those who were less active.21 Other lifestyle changes or factors that can decrease the risk of Alzheimer’s disease include following a Mediterranean diet, consumption of coldwater fish and omega-3 diets, avoidance of head trauma, and not smoking.22 Anticholinergic drugs and proton pump inhibitor drugs have been shown to be associated with increased risk of Alzheimer’s disease.23,24 Environmental exposures such as second-hand smoke, air pollution and pesticides have also been shown to possibly increase the incidence of Alzheimer’s disease.25
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
Similarly to Alzheimer’s disease, the exact etiology of Parkinson’s disease is not known, but the disease involves an age-related progressive loss of neurons. Parkinson’s disease initially manifests with resting tremor due to destruction of the dopaminergic neurons in the nigrostriatal pathway (substantia nigra pars compacta of the midbrain). The incidence of Parkinson’s disease is estimated to be about 0.3% in individuals over age 40, and it is estimated that 7.5 million people worldwide have Parkinson’s disease.26 The risk factors for Parkinson’s disease are similar to those for Alzheimer’s disease, except that in addition to family history, genetics, age, diet, and exercise, other factors, such as increased exposure to pesticides and other chemicals and a possible link to dairy products, appear to increase the risk.27
In 1912, Lewy showed that there were neuronal cytoplasmic inclusion bodies in Parkinson’s disease that we now know contain large amounts of α-synuclein. As Parkinson’s disease progresses, however, other brain areas become involved, exhibiting a widespread loss of neurons, dementia characterized by Lewy bodies throughout the brain, and symptoms that include memory loss and cognitive decline. The principal component of the Lewy bodies is aggregates (oligomers) of α-synuclein,28 a protein that is abundant in neurons and localized to the synaptic region. The ability of α-synuclein to be transferred to other cells may explain why it is possible for “seeding” or propagation of misfolded proteins to occur.29 The formation of α-synuclein aggregates precedes cell death, and mutations in α-synuclein lead to early-onset Parkinson’s disease and oligomer formation.30 Similarly to Alzheimer’s disease, oxidative stress, reactive oxygen species, and mitochondrial dysfunction are theorized to be the most prominent explanations of the cause of the disease. Reactive oxygen species generated from dopamine precursors and breakdown products are thought to be the major causes of dopaminergic cell death.31
RECENT ADVANCES IN EARLY-ONSET PARKINSON’S DISEASE GENETICSAdvances in the genetics of Parkinson’s disease point toward the autophagy pathway as critical for neuronal survival and as at least partially responsible for neurodegeneration. Autosomal recessive mutations in the phosphatase and tensin homolog–induced putative kinase 1 (PINK1) gene have been shown to induce a neurodegeneration in people that is similar to Parkinson’s disease, and PINK1 is the second most common genetic cause of early-onset Parkinson’s disease (after Parkin).32 The Parkin gene, which has been shown to be the major cause of early-onset (familial) Parkinson’s disease and a major risk factor in the more common sporadic form, also may contribute to Alzheimer’s disease.33PINK1 encodes a kinase that labels damaged mitochondria for destruction by autophagy. PINK1 recruits Parkin (a ubiquitin ligase) to label not only mitochondria, but also damaged proteins and proteins aggregates for autophagy.34 This evidence suggests that major genetic causes and influences of both early-onset Alzheimer’s disease and Parkinson’s disease are linked to autophagy. Also, dysregulation of autophagy in the substantia nigra of patients with Parkinson’s disease has been demonstrated.35 Another gene product, called F-box protein 7, has been linked to juvenile-onset Parkinson’s disease with atypical features including dementia, dystonia, hyperreflexia and pyramidal effects.36 F-box protein recruits Parkin to damaged mitochondria and mutants fail to recruit damaged mitochondria for autophagy.37
MOLECULAR TARGET OF RAPAMYCIN PATHWAY AND AUTOPHAGYPerhaps the most convincing clinical evidence for the role of autophagy in neurodegenerative disease comes from the recent support for the effect of rapamycin on the molecular target of rapamycin (mTOR) pathway. The mTOR pathway (mTOR is a Ser/Thr kinase that is activated by phosphoinositide 3-kinase [PI3K] growth factors) promotes multiple functions in the cell. mTOR complex 1 (mTORC1) controls protein homeostasis and autophagy, and it can also prevent apoptosis, particularly in neurons.37 If mTORC1 activity is ablated, neurons undergo apoptosis and death due to oxidative stress.38,39 The mTORC2 pathway, which also responds to PI3K activation by growth factors, primarily regulates the cell cytoskeleton, cell survival, and metabolism.37 Loss of mTOR signaling can impair long-term potentiation and synaptic plasticity in animal models of AD that can be restored by mTOR activation.40
The regulation of autophagy by mTORC1 is most relevant for the survival and regeneration of neurons and for the effect on neurodegenerative diseases. Basal levels of autophagy are required for neuronal survival and function.41 Autophagy prevents the buildup of toxic protein aggregates and also is critical for disposal of dysfunctional mitochondria. Autophagy involves targeting misfolded proteins, protein aggregates, and dysfunctional organelles (such as mitochondria) to lysosomes. Molecular chaperones are also critical in targeting both protein aggregates and dysfunctional mitochondria to lysosomes (the Parkin complex is a molecular chaperone for lysosomal targeting). Rapamycin (a partial inhibitor of mTOR at low doses) enhances lysosomal degradation, reduces protein aggregation, and prevents cognitive deterioration in mice with Alzheimer’s disease because it increases autophagy.42 Further studies have shown that low-dose rapamycin can decrease the tau hyperphosphorylation and tau cortical tangles43 that are characteristic of Alzheimer’s disease. In an extremely interesting experiment, rapamycin abrogated tau hyperphosphorylation in neurons even with a high-sucrose diet, despite the fact that hyperinsulinemia occurred in the periphery.44 This is most likely due to the fact that mTOR signaling is downstream from insulin receptor (and growth factor) signaling (and after PI3K signaling). mTOR signaling appears to be the “master regulator” of autophagy and inhibits autophagy by the inhibition transcription factor EB (TFEB) that stimulates autophagy-related genes to turn on autophagy, as well as by inhibition of UNC-51–like kinase.
Additionally, many natural substances have been shown to enhance autophagy in either an mTOR-dependent or mTOR-independent manner. Some of the effects of natural substances on neurodegeneration may be as antioxidants (inhibiting protein aggregation or cellular damage), as inhibitors of inflammation and autophagy (see Figure 2, TNF-α inhibits autophagy), enhancing molecular chaperones such as heat shock protein (curcumin),45 or acting directly as mTOR-dependent or mTOR-independent (trehalose) inhibitors of autophagy.46 Additionally, other drugs and substances can increase autophagy by inhibition of phosphoinositol signaling (lithium) and calpain inhibition.
Some phytonutrients may have multiple mechanisms of action in neurodegenerative diseases. For instance, quercetin has been shown to have a direct antioxidant effect, activate Nrf2 (and decrease intracellular oxidative damage), activate sirtuins (and activating the stress response) and increase autophagy.46 There is some linkage between the autophagy and Nrf2 pathways that control, respectively, protein misfolding and intracellular oxidative stress because activation of Nrf2 also indirectly activates autophagy.
CURRENT THEORIES OF NEURODEGENERATION IN ALZHEIMER’S DISEASEGiven the pathological features and the molecular and genetic clues along with the epidemiological risk factors, a number of theories have been proposed to explain the pathophysiology of plaque formation. The three main theories on the cause and progression of Alzheimer’s disease are inflammation in the CNS, mitochondrial dysfunction and reactive oxygen species damage that accumulates over time, leading to the plaques and neurofibrillary tangles found Alzheimer’s disease. These plaques and tangles and the protein aggregates contribute to neuronal apoptosis and other brain cell death and necrosis. These three theories postulate that epigenetic environmental factors contribute to Alzheimer’s disease by altering the function or survival of neurons. However, the ability of neurons or microglia to decrease protein aggregates is actually affected by inflammation, reactive oxygen species and inflammation. Newer research on autophagy (see below, based on the original work of Alois Alzheimer) pathology identified neurofibrillary tangles and plaques as possible causative agents of Alzheimer’s disease, now allows integration and unification of following the main neurodegenerative disease theories.
INFLAMMATION THEORY OF NEURODEGENERATIONThe first theory is the inflammation hypothesis for neurodegenerative disease. APP (the amyloid precursor protein that affects β-catenin membrane anchoring to the actin cytoskeleton) is cleaved by γ-secretase initially (activated by presenilins 1 and 2)47 and then into Aβ42 (a proinflammatory fragment that can form aggregates). Inhibitors of the inflammatory cytokine TNF-α prevent the cleavage and accumulation of Aβ42 and the cognitive defects of Alzheimer’s disease in mice.48 There is newer evidence, however, that Aβ secretion and plaque formation are fundamentally dependent on autophagy and that autophagy can remarkably reduce extracellular plaque burden.49 Induction of autophagy by rapamycin lowers plaque burden,50 and oxidative stress reduces autophagy and increases Aβ generation.51 Aβ aggregates also stimulate the microglial (macrophage-like) cells in the brain to become activated (to the M1 inflammatory phenotype), and M1 microglial cells in turn promote further inflammation by secreting the inflammatory cytokine TNF-α.52 Microglia are also capable of phagocytosing and digesting protein aggregates, so the specific type of activation of microglia becomes critical to disease progression. In general, an inflammatory state promotes increased inflammatory cytokines (interleukin [IL]-1β and TNF-α), and these cytokines increase Aβ aggregates, tau phosphorylation and aggregates, and disease progression. Some have hypothesized that Aβ leads to tau aggregates, whereas others have posited that tau aggregates start first and lead to Aβ aggregates.53 Experiments with mice have shown that injecting abnormal fibrillar aggregates of Aβ can lead to the spread of disease to normal proteins, possibly by “seeding” similar to prion “seeding” of abnormal proteins.54 Rapamycin (increases autophagy at low doses, partial inhibitor of mTOR) significantly reduces cortical tangles of tau, decreases tau hyperphosphorylation and lowers the levels of tau in the mouse forebrain.43 Taken together, partial stimulation of autophagy can prevent both tau and Aβ accumulation (the pathological hallmarks of Alzheimer’s disease) and the symptoms of Alzheimer’s disease.
In addition to the effects of inflammation on Aβ, the hyperphosphorylation of tau is also partially regulated by inflammation. In mice with tau hyperphosphorylation and tau aggregation, TREM2 can be overexpressed in microglia, and overexpression of TREM2 on microglia rescued spatial cognitive impairments and prevented neuronal and synaptic loss as well as tau hyperphosphorylation.55 TREM2 also promotes the anti-inflammatory M2 phenotype in glial (macrophage-like) cells, demonstrating that decreased inflammation ameliorates Alzheimer’s disease symptoms and tau aggregation. Inflammation both prevents clearance of tau and neurofibrillary tangles and generates Aβ fragments that promote plaques. However, clearance by microglial cells occurs in either already damaged cells or outside cells and is secondary to the initial accumulation of protein aggregates. Also, Aβ aggregates induce inflammation, so the primary defect is the production of these protein aggregates and factors that decrease neural inflammation ameliorate Alzheimer’s disease.
MITOCHONDRIAL DYSFUNCTION THEORY OF NEURODEGENERATIONThe second theory, the mitochondrial dysfunction theory, postulates that altered mitochondrial function (dysfunctional mitochondria) causes or exacerbates Alzheimer’s disease. Protein aggregates of Aβ may damage mitochondria, or mitochondrial damage might precede protein aggregation. It is clear that mitochondrial dysfunction occurs early in Alzheimer’s disease. Lower percentages of mitochondria, shorter and wider mitochondria, decreased axonal transport of mitochondria and impaired electron transfer, disruption of the glycolytic process, and decreased antioxidant activity have all been shown in Alzheimer’s disease.56 Disruption of mitochondrial function is a well-described pathological process that leads to decreased adenosine triphosphate, cell swelling, and eventual cell death by necrosis or apoptosis (cytochrome c release). Aβ toxicity to neurons has been theorized to be partially due to mitochondrial damage.57 One of the major ways that cells dispose of and clear malfunctioning mitochondria is by autophagy (also termed mitophagy). An accumulation of damaged, malfunctioning mitochondria in neurons can trigger apoptosis or necrosis in neurons. Dysfunctional, enlarged, senescent mitochondria accumulate during the aging process and also as Alzheimer’s disease progresses. These dysfunctional mitochondria cannot fuse together (a normal stress response to nutrient deprivation) and cannot signal autophagy.58 Mitochondria also help regulate autophagy and the sirtuin1 gene product also promotes autophagy.58
OXIDATIVE STRESS/REACTIVE OXYGEN THEORY OF NEURODEGENERATIONThe third theory is that reactive oxygen species and a lack of Nrf2 activation of cellular defenses lead to increased protein damage (in addition to cell membrane and DNA damage). Damaged proteins, particularly amyloid protein, aggregate into fibrils, and amyloid fibrils are toxic to neurons, astrocytes, and other cells, leading to neuronal death.59 Protein aggregates are removed from cells primarily by autophagy and targeting to lysosomes.
Oxidative stress and generation of reactive oxygen species have been postulated to be important contributors to the progression of Alzheimer’s disease. Clearly, oxidative damage to intracellular proteins can initiate protein misfolding and aggregation due to exposure of hydrophobic residues. Additionally, epidemiological evidence indicates that diet and lifestyle alter the predisposition to and risk of developing Alzheimer’s disease. Cell damage and inflammation initiated by reactive oxygen species contribute to activating inflammation and M1 microglial cells. M1 activation by TNF-α and IL-1β inflammatory cytokines increases plaque formation and promotes Alzheimer’s disease. Extensive reports show that free radicals are pathologically critical in Alzheimer’s disease.60 One theory that has been proposed is that oxidized cholesterol may contribute to Alzheimer’s disease. The APOɛ4 allele (a major risk factor for Alzheimer’s disease) has been implicated in generating oxidative stress and also in mitochondrial dysfunction.61 APOɛ4 also appears to induce increased inflammation and inflammatory cytokine release in microglial and astrocytes cells.62 In summary, oxidized cholesterol probably contributes to hypertension and inflammation in the brain, and both of these factors contribute to Alzheimer’s disease.
GENETIC CAUSES OF EARLY ONSET AND DECREASED AUTOPHAGY IN PARKINSON’S DISEASEThere are more direct genetic links to early-onset Parkinson’s disease that yield clues to the underlying causes. In Parkinson’s disease, genetic defects in SNCA (α-synuclein), Parkin, PINK1, OMI (Park13), and leucine-rich repeat Ser/Thr kinase 2 (LRRK2) cause early-onset Parkinson’s disease, and all these genetic defects point toward defects in both autophagy and mitochondrial function.63 The major α-synuclein (Park1) mutations lead to protein aggregation and overexpression of α-synuclein mutants, cause mitochondrial degeneration,64 and lead to symptoms of Parkinson’s disease. The LRRK2 mutations (also localized to the mitochondria) lead to mitochondrial dysfunction,62 and inhibition of LRRK2 leads to autophagy that is independent of mTOR but dependent on Beclin-2.63 (Beclin-2 is inhibited by PI3K.) An mTOR- and rapamycin-independent pathway for induction of autophagy via Beclin-2 is important for the mTOR-independent mechanisms of some natural substances that induce autophagy (see Table 1). Parkin (Park2) was the first recessive gene associated with Parkinson’s disease, and mutations in Parkin lead to an early-onset recessive juvenile disease.82 Parkin is a ubiquitin ligase that tags proteins and organelles for destruction. Recent evidence shows that Parkin is essential for mitophagy and that Parkin-mediated ubiquitination of mitochondrial proteins triggers degradation of dysfunctional mitochondria through the autophagy-lysosome pathway.83 PINK1 is also a mitochondrial Ser/Thr kinase, recruits Parkin to the mitochondria, and is critical for mitophagy (autophagy of mitochondria).84 Another ubiquitin ligase (similar to Parkin) is F-box protein 7. It also plays a role in mitophagy, but Parkin can be recruited to impaired mitochondria, whereas F-box protein 7 is induced by cellular stress and can recruit Parkin to damaged mitochondria.85 In summary, most of the genetic mutations in Parkinson’s disease effect either mitochondrial dysfunction or autophagy/mitophagy. The PINK1 and Parkin genes are also activated or regulated by mTOR and rapamycin (a macrolide antibiotic used as an immunosuppressant that binds to an intracellular protein called FK506).86 Rapamycin has been shown to inhibit mTOR, enhance autophagy (at low doses), reduce loss of neurons, protect from oxidative stress in mice, prevent apoptosis and improve Parkinson’s symptoms.87
Table 1: Summary of natural substances and their effect on autophagy and inflammation.
| Natural Substance | Mechanism of Action | Reference |
|---|---|---|
| Sulforaphane | mTOR dependent increased autophagy, Nrf2 activation |
65 |
| Arctigenin | Increases autophagy | 66 |
| Resveratrol | Heme-oxygenase–dependent increase in autophagy Nrf2 activation |
67–69 |
| Trehalose | mTOR-independent increase in autophagy | 70 |
| Epigallocatechin | Increases autophagy Nrf2 activation |
71 |
| Tripchlorolide/triptolide | mTOR dependent increased autophagy | 72, 73 |
| Curcumin | mTOR dependent increased autophagy Activates TFEB HSP chaperones |
74 |
| Quercetin | mTOR activation Nrf2 activation |
75 |
| Vitamin E | Reduced vitamin level reduces autophagy | 76 |
| Conophyllin | mTOR dependent autophagy in Huntington’s disease | 77 |
| Berberine | Activates Akt and mTOR Activates Nrf2 |
78 |
| Oleocanthol, oleuropein (olive oil) | Oleocanthol increases clearance of protein aggregates Oleuropein activates mTOR and AMPK, increases autophagy |
79, 80 |
| Coenzyme Q10 | Antioxidant and mitochondrial effect | 81 |
AMPK, Adenosine monophosphate-activated protein kinase; HSP, heat shock protein; mTOR, molecular target of rapamycin; TFEB, transcription factor EB.
INFLAMMATION AND PARKINSON’S DISEASEIn addition to the strong evidence that links the causes of Parkinson’s disease to impaired autophagy, there is additional evidence that, similarly to in Alzheimer’s disease, inflammation and microglial activation can contribute to Parkinson’s disease. Inflammation also plays a key role in the damage to dopaminergic neurons in Parkinson’s disease (similarly to Alzheimer’s disease), and microglia can be protective or induce further inflammation. Specifically, microglia secrete anti-inflammatory cytokines such as transforming growth factor-β, IL-10, IL-13, and IL-4 (M2-type secretion) that decrease inflammation and help clear protein aggregates in Parkinson’s disease or they can secrete inflammatory cytokines such as TNF-α that exacerbate Parkinson’s disease.88 In fact, recent evidence shows that inflammatory cytokines such as TNF-α secreted from microglial cells can actually reduce α-synuclein degradation by suppressing autophagy in dopaminergic neurons.89 The key take-home message from this research is that inflammation is not just a result of neuronal damage; it also enhances neuronal damage by inhibition of autophagy.
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
It is now becoming apparent that a number of neurodegenerative diseases besides Alzheimer’s disease and Parkinson’s disease are also primarily affected by mitochondrial dysfunction and impaired autophagy. Specifically, there is evidence that Huntington’s disease and amyotrophic lateral sclerosis (ALS) also have defects in autophagy and that rapamycin can reverse Huntington’s disease and trehalose, resveratrol and lithium can reverse ALS.54
Neurodegeneration and cell death in general may also be implicated in aging. A number of lines of research indicate that defective autophagy is critical in aging. Perhaps the most convincing evidence again comes from studies of rapamycin. Although there have been only a few studies of low-dose rapamycin in humans, researchers in a recent pilot study looked at markers of aging in people taking low-dose rapamycin after cardiac rehabilitation and saw improvement in senility markers such as TNF-α and β-galactosidase.90 In cellular studies, low-dose rapamycin restores autophagy, decreases mitochondrial dysfunction, and blocks senescence-related changes in cells.91 In animal models, low-dose rapamycin has been shown to prolong lifespan (and make animals healthier) in organisms as diverse as yeast, mice and mammals. Interestingly, caloric restriction (one of the few other proven antiaging treatments) also inhibits mTORC1.92 On one hand, insufficient autophagy causing accumulation of protein aggregates and the failure to recycle mitochondria lead to toxic buildup of protein aggregates; additionally, the failure to recycle mitochondria leads to increased reactive oxygen species generation and additional buildup of protein aggregates. On the other hand, too much autophagy (higher-dose rapamycin) can also lead to cell death, whereby the cell “eats itself.” The balancing point of partial autophagy appears to be best for cell survival. Partial inhibition of autophagy by low-dose rapamycin, short-acting phytonutrients, or partial nutrient deprivation can activate sufficient autophagy to eliminate dysfunctional mitochondria and prevent the buildup of protein aggregates (Figure 1). Preventing apoptosis may not be enough to prevent neuronal cell death, because protein aggregates can cause necrosis and/or necroptosis. Neuronal survival factors such as brain-derived neurotrophic factor (BDNF) or nerve growth factor may prevent neurons from undergoing apoptosis, but neurons can still die as a result of toxic buildup of protein aggregates or the inability to dispose of dysfunctional mitochondria. Mitochondria that are dysfunctional can also contribute to further oxidative damage by releasing free radical species, or they can directly trigger apoptosis by opening the mitochondrial pore (Bax/Bac channel) and releasing cytochrome c. It is the balance between too much autophagy that can lead to cell death and too little autophagy that can lead to protein accumulation and dysfunctional mitochondria that also leads to cell death that is critical. Healthy, long-lived neurons require enough autophagy to get rid of the toxic buildup of protein aggregates and dysfunctional proteins, but not too much that leads to complete autophagy and cell death (Figure 2). If protein aggregates build up inside neurons, these aggregates can “seed” to other neurons, accumulate in the extracellular space, and trigger phagocytosis and activation of microglial cells (leading to increased M1 phenotype as well as IL-1 TNF-α secretion). Increased inflammation in turn can lead to decreased autophagy and more protein aggregation as well as increased cell death (Figure 2). The cycle of cell death, inflammation, and mitochondrial dysfunction can be broken by increased autophagy and decreased inflammation. Natural substances that can act both as antiinflammatory agents and as partial inhibitors of autophagy may have particular use in the prevention and treatment of neurodegenerative disorders. Also, targeting different mechanisms (mTOR-dependent and mTOR-independent pathways) with multiple autophagy stimulators at the lower doses found in fruits and vegetables may be a more effective technique than trying to use a single lower-dose drug to stimulate autophagy and preserve neurons.
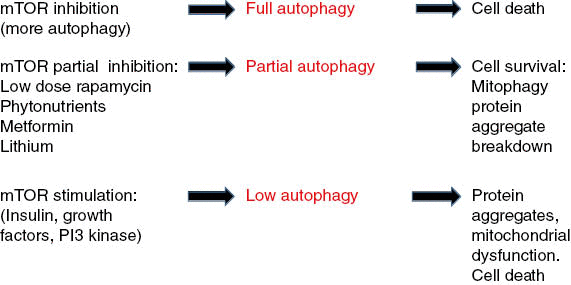
Figure 1: Full inhibition of molecular target of rapamycin (mTOR; high-dose rapamycin) stimulates full autophagy that leads to eventual cell death.
Partial inhibition of mTOR (resveratrol, other phytonutrients, metformin, adenosine monophosphate–activated protein kinase–stimulation) causes partial autophagy, targeting of misfolded proteins, and dysfunctional mitochondria to lysosomes (mitophagy). Full stimulation of mTOR causes inhibition of autophagy (insulin, nutrient excess, phosphoinositide 3-kinase activation) leads to protein aggregate accumulations and dysfunctional mitochondria (lack of mitophagy) and eventual cell death seen in neurodegenerative disease.
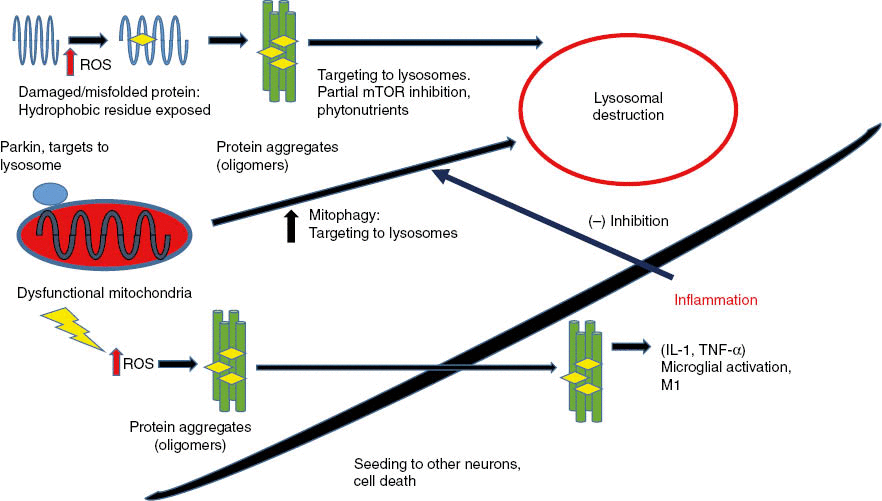
Figure 2: Misfolded proteins, damaged by reactive oxygen species (ROS), form insoluble aggregates in neurons that trigger neuronal cell death.
Dysfunctional mitochondria generate ROS that damage proteins and cause protein aggregation. Partial autophagy activates lysosomal degradation that degrades both protein aggregates and dysfunctional mitochondria. When protein aggregates build up inside neurons, they trigger cell death by apoptosis. Protein aggregates can “seed” and spread to adjacent neurons similarly to prions. As more neurons die, protein aggregates accumulate outside cells, are phagocytosed, and activate microglial cells to secrete inflammatory cytokines (TNF-α and IL-1). Inflammation signals outside neurons then inhibit autophagy and promote increased neuronal death.
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
Whereas rapamycin is already approved by the US Food and Drug Administration as a drug for immunosuppression and renal cell carcinoma, it is not approved for neurodegenerative disorders, and it has major adverse effects at higher doses (lower doses have been used for most of the autophagy experiments in human and mice). Currently, there are clinical trials of rapamycin only in dogs (volunteered pets), but not yet in humans, looking at canine longevity.93 However, there is abundant evidence that many natural substances can alter CNS inflammation, alter autophagy and have neuroprotective properties. In attempts to classify the research into the effects of natural substances on neurodegenerative disorders, the natural substances will be separated by mechanism of action including mTOR-dependent and mTOR-independent effects on autophagy, effects on neuroinflammation and microglial cells, mitochondrial dysfunction, enhancement of neurotrophins (neuronal survival factors) such as BDNF, reactive oxygen species inhibition, and other natural substances with unknown mechanisms but positive effects on neurodegenerative disease. Some natural substances may have multiple mechanisms of action but are classified below according to their presumed primary mechanisms.
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
SULFORAPHANESulforaphane is a naturally occurring substance in cruciferous vegetables. In vitro evidence has shown that sulforaphane can be cytoprotective in chemical damage to neurons.94In vivo sulforaphane has been shown to protect mice from damage to the nigrostriatal dopaminergic neurons.95 Further elucidation of sulforaphane’s mechanisms of action demonstrates that its protection against neurodegeneration in mice (rotenone-induced) is due to stimulation of autophagy (mTOR-dependent) and Nrf2 activation.65 Interestingly, sulforaphane may inhibit reactive oxygen species by activating intracellular defense mechanisms with Nrf2 (decreasing the amount of misfolded protein), and it may also enhance autophagy to remove misfolded protein aggregates and dysfunctional mitochondria. However, this study did not assess mitochondrial function, but it did demonstrate that sulforaphane can be protective in Parkinson’s disease and probably in Alzheimer’s disease as well. (Although this was not tested directly, enhancement of autophagy would be beneficial.)
ARCTIGENINArctigenin (a lignin found in plants of the Asteraceae, including the greater burdock [Arctium lappa]) reduces memory impairment in mice, reduces Aβ aggregates and enhances autophagy in both human cells and mice.66
RESVERATROLResveratrol (a stilbenoid or natural phenol) is found mostly in the skin of grapes (and in red wine), blueberries, and raspberries. Concentrations of resveratrol are increased when berry or grape plants are exposed to fungi. Resveratrol has been shown to increase autophagy in human cells in a heme oxidase–dependent manner and to increase mitochondrial function.67,68 Resveratrol has also been shown to induce Nrf2 (and activate intracellular antioxidation defenses) and reduce oxidative stress in rats.69
TREHALOSETrehalose is a disaccharide found in bacteria, yeast, fungi, insects, plants, and invertebrates but it is not endogenously synthesized in mammals. Trehalose has been shown to inhibit amyloid aggregation and clear α-synuclein and mutant Huntingtin proteins by an mTOR-independent enhancement of autophagy.70 Because trehalose induces autophagy in an mTOR-independent manner, it could be used synergistically with an mTOR-dependent autophagy inducer such as sulforaphane.
EPIGALLOCATECHINEpigallocatechin is found in high amounts in green tea, and it enhances autophagy in macrophages; however, the mechanism is not clear.71
TRIPCHLOROLIDE AND TRIPTOLIDEBoth these compounds are derived from Tripterygium wilfordii, a Chinese herb that has antiinflammatory properties. Tripchlorolide improves cognitive deficits, has been shown to be neuroprotective in Parkinson’s disease, and activates the mTOR pathway.72 Triptolide (the better-studied nuclear factor-κB inhibitor constituent) induced autophagy in human PC12 cells in an Alzheimer’s disease model.73
CURCUMINCurcumin (a polyphenol, and the active constituent in turmeric) is derived from the Curcuma longa plant. In rodent models, curcumin reduced plaque burden, protected against Aβ toxicity, and improved cognitive function.96 Curcumin enhances autophagy by an mTOR-mediated pathway, activates a TFEB-lysosome pathway, and enhances heat shock protein chaperones.74 Curcumin may be useful for all of the neurodegenerative diseases where autophagy is reduced.
QUERCETINQuercetin is a flavonol that is commonly found in fruits and vegetables (apples, berries and onions have some of the higher amounts of quercetin) as a quercetin glycoside. (Note that quercetin in supplements is quercetin aglycone.) Quercetin was shown to decrease cognitive deficits in a mouse model of Alzheimer’s disease and to reduce Aβ aggregates.97 Interestingly, coffee has been shown to decrease the risk of both Alzheimer’s disease and Parkinson’s disease, and authors of a recent paper postulated that quercetin, not caffeine, is the major neuroprotective constituent in coffee.98 Quercetin has been shown to activate mTOR and Nrf2, but there are still no direct data yet on the effect of quercetin on autophagy in neurodegenerative diseases.75
VITAMIN EThe α-, γ- and δ-forms of tocopherol have all been found to reduce Aβ formation in cultured cells, but δ-tocopherol was the most potent.99 In an interesting experiment, long-term vitamin E–deficient mice had reduced autophagy.76
CONOPHYLLINConophylline is a vinca alkaloid isolated from the tropical plant Tabernaemontana divaricata. Conophylline can induce an mTOR-dependent autophagy in neuronal cells and eliminated Huntingtin protein aggregates.77
BERBERINEBerberine is an alkaloid found in the roots of plants such as Berberis vulgaris (barberry), Berberis aristata (tree turmeric), Mahonia aquifolium (Oregon grape), Hydrastis canadensis (goldenseal), Xanthorhiza simplicissima (yellowroot), and Coptis chinensis (Chinese goldthread). Berberine has been used to counter the effects of type 2 diabetes. Researchers in a recent study found that berberine can attenuate glucose neurotoxicity and activate Nrf2 and Akt (mTOR pathway).78 Berberine also prevented protein aggregation in an ALS model in an mTOR-dependent manner.100
OLEOCANTHOL AND OLEUROPEIN (FROM OLIVE OIL)Olive oil is an important component of the Mediterranean diet that has been shown to decrease the risk of Alzheimer’s disease. Oleuropein aglycone is the main polyphenol found in extra virgin olive oil. Oleuropein enhances autophagy by an mTOR- and adenosine monophosphate–activated protein kinase–dependent mechanism.79
There are about 36 different phenolic compounds in olive oil, but oleocanthol is one of the more common phenols, and it has been shown have antiinflammatory effects. In Alzheimer’s disease models, oleocanthol has been shown to enhance the clearance of Aβ aggregates. The mechanism for this was postulated to be upregulation of proteolysis and clearance, but no data on the effect on autophagy were reported.80
COENZYNME Q10 (CoQ10)Coenzyme Q10 (CoQ10; ubiquinol, the nonoxidized form) is a naturally occurring compound that is normally found in cells as part of the electron transport chain in mitochondria. The mechanism of action of CoQ10 is presumed to be as a strong antioxidant or as an enhancer of mitochondrial function. A recent paper showed that CoQ10 can enhance autophagy in retinal cells.81 A recent meta-analysis of the effect on CoQ10 in Parkinson’s disease showed that CoQ10 was not superior to placebo for treatment of Parkinson’s symptoms.101 This does not mean, however, that CoQ10 might have some preventive effects in early Parkinson’s disease.
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
There is a strong relationship between autophagy and inflammatory processes. Chronic stress and chronic inflammation can lead to increased signaling for apoptosis and inhibit autophagy. Additionally, autophagy can reduce dysfunctional mitochondria and reactive oxygen species generation as well as decrease protein aggregate accumulations. Genetic defects in autophagy lead to neuropathological accumulation of damaged mitochondria and protein aggregates that can manifest in early-onset Alzheimer’s disease and Parkinson’s disease. These genetic clues point to the process of autophagy as a critical step in neuropreservation. Partial activation of autophagy is key to neuronal health. Insufficient autophagy can lead to accumulation of protein aggregates and dysfunctional mitochondria. Too much autophagy results in cell death. Exercise, caloric restriction, and a diet rich in fruits and vegetables partially activate autophagy and are thus important epidemiological factors that may help prevent cognitive decline symptomatic of neurodegenerative disease. Natural substances that stimulate partial autophagy through mTOR-dependent or mTOR-independent pathways decrease protein aggregates and ameliorate neurodegenerative disease. The paradigm shift toward lifestyle modification, drugs, and natural substances that enhance partial autophagy is already yielding scientific evidence and possible new treatments for Alzheimer’s disease and Parkinson’s disease.
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
The author declares he has no competing interests.
- TOP
- ABSTRACT
- INTRODUCTION
- ALZHEIMER’S DISEASE
- PARKINSON’S DISEASE
- NEURODEGENERATIVE DISEASES, PARTIAL AUTOPHAGY, AND AGING
- NATURAL SUBSTANCES AND NEURODEGENERATIVE DISEASE
- NATURAL SUBSTANCES THAT ENHANCE AUTOPHAGY (FOR PREVENTION OR TREATMENT OF NEURODEGENERATIVE DISEASE)
- CONCLUSIONS
- COMPETING INTERESTS
- REFERENCES
- Jackson MP,
Hewitt EW.
Cellular proteostasis: degradation of misfolded proteins by lysosomes.
- Wang B,
Abraham N,
Gao G,
Yang Q.
Dysregulation of autophagy and mitochondrial dysfunction in Parkinson’s disease.
- Castellani RJ,
Zhu X,
Lee HG, et al. Molecular pathogenesis of Alzheimer’s disease: reductionist versus expansionist approaches.
- Hebert LE,
Weuve J,
Scherr PA,
Evans DA.
Alzheimer disease in the United States (2010–2050) estimated using the 2010 census.
- Prince M,
Bryce R,
Albanese E, et al. The global prevalence of dementia: a systematic review and meta-analysis.
- Sosa-Ortiz AL,
Acosta-Castillo I,
Prince MJ.
Epidemiology of dementias and Alzheimer’s disease.
- Braak H,
Braak E.
Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections.
- Hyman BT,
Phelps CH,
Beach TG, et al.
National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease.
- Larson EB,
Yaffe K,
Langa KM.
New insights into the dementia epidemic.
- Farrer LA,
Cupples LA,
Haines JL, et al.
Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis.
- van Duijn CM,
Clayton D,
Chandra V, et al. Familial aggregation of Alzheimer’s disease and related disorders: a collaborative re-analysis of case-control studies.
- Guerreiro R,
Wojtas A,
Bras J, et al. TREM2 variants in Alzheimer’s disease.
- Helzner EP,
Luchsinger JA,
Scarmeas N, et al. Contribution of vascular risk factors to the progression in Alzheimer disease.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial.
- Honig LS,
Kukull W,
Mayeux R.
Atherosclerosis and AD: analysis of data from the US National Alzheimer’s Coordinating Center.
- Költringer P,
Langsteger W,
Eber O.
Dose-dependent hemorheological effects and microcirculatory modifications following intravenous administration of Ginkgo biloba special extract EGb 761.
- Whitmer RA,
Sidney S,
Selby J, et al.
Midlife cardiovascular risk factors and risk of dementia in late life.
- Whitmer RA.
The epidemiology of adiposity and dementia.
- Martin-Jiménez C,
Gaitán-Vaca D,
Echeverria V, et al.
Relationship between obesity, Alzheimer’s disease and Parkinson’s disease: an astrocentric view.
doi: https://doi.org/10.1007/s12035-016-0193-8.
PubMed PMID: 27796748 . - Profenno L,
Porsteinsson A,
Faraone S.
Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders.
- Hamer M,
Chida Y.
Physical activity and risk of neurodegenerative disease: a systematic review of prospective evidence.
- Chouliaras L,
Sierksma AS,
Kenis G, et al.
Erratum: gene-environment interaction research and transgenic mouse models of Alzheimer’s disease.
- Gray L,
Anderson L,
Dublin S, et al.
Cumulative use of strong anticholinergics and incident dementia: a prospective cohort study.
- Gomm W,
von Holt K,
Thomé F, et al.
Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis.
- Richardson J,
Roy A,
Shalat S, et al.
Elevated serum pesticide levels and risk for Alzheimer disease.
- Pringsheim T,
Jette N,
Frolkis A,
Steeves TD.
The prevalence of Parkinson’s disease: a systematic review and meta-analysis.
- Noyce J,
Bestwick P,
Silveira-Moriyama L, et al.
Meta-analysis of early nonmotor features and risk factors for Parkinson disease.
- Jakes R,
Spillantini G,
Goedert M.
Identification of two distinct synucleins from human brain.
- Hansen C,
Angot E,
Bergström L, et al.
α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells.
- Houlden H,
Singleton B.
The genetics and neuropathology of Parkinson’s disease.
- Perfeito R,
Cunha-Oliveira T,
Rego AC.
Reprint of: revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease – resemblance to the effect of amphetamine drugs of abuse.
- Arena G,
Valente EM.
PINK1 in the limelight: multiple functions of an eclectic protein in human health and disease.
- Zhang CW,
Hang L,
Yao TP,
Lim KL.
Parkin regulation and neurodegenerative disorders.
- Michiorri S,
Gelmetti V,
Giarda E, et al.
The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy.
- Nixon RA.
The role of autophagy in neurodegenerative disease.
- Deng H,
Liang H,
Jankovic J.
F-box only protein 7 gene in parkinsonian-pyramidal disease.
- Lan AP,
Chen J,
Zhao Y, et al.
mTOR signaling in Parkinson’s disease.
- Chen L,
Xu B,
Liu L, et al.
Hydrogen peroxide inhibits mTOR signaling by activation of AMPKα leading to apoptosis of neuronal cells.
- Choi KC,
Kim SH,
Ha JY, et al.
A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death.
- Ma T,
Hoeffer CA,
Capetillo-Zarate E, et al.
Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease.
- Hara T,
Nakamura K,
Matsui M, et al.
Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice.
- Ravikumar B,
Duden R,
Rubensztein D.
Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy.
- Ozcelik S,
Fraser G,
Castets P, et al.
Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice.
- Orr ME,
Salinas A,
Buffenstein R,
Oddo S.
Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer’s disease pathology.
- Maiti P,
Manna J,
Veleri S,
Frautschy S.
Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin.
doi: https://doi.org/10.1155/2014/495091. Epub 2014 Oct 19 . - Costa LG,
Garrick JM,
Roquè PJ,
Pellacan C.
Mechanisms of neuroprotection by quercetin: counteracting oxidative stress and more.
doi: https://doi.org/10.1155/2016/2986796. Epub 2016 Jan 24 . - Nizzari M,
Venezia V,
Repetto E, et al.
Amyloid precursor protein and Presenilin1 interact with the adaptor GRB2 and modulate ERK 1,2 signaling.
- Detrait E,
Danis B,
Lamberty Y,
Foerch P.
Peripheral administration of an anti-TNF-α receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-α levels and memory deficits in mice.
- Nilsson P,
Loganathan K,
Sekiguchi M, et al.
Aβ secretion and plaque formation depend on autophagy.
- Yu H,
Cuervo M,
Kumar A, et al.
Macroautophagy – a novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease.
- Zheng L,
Terman A,
Hallbeck M, et al.
Macroautophagy-generated increase of lysosomal amyloid β-protein mediates oxidant-induced apoptosis of cultured neuroblastoma cells. Autophagy.
2011; 7:1528–45. - Birch AM,
Katsouri L,
Sastre M.
Modulation of inflammation in transgenic models of Alzheimer’s disease.
- Jack CR Jr,
Knopman DS,
Jagust WJ, et al.
Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers.
- Ashe KH,
Aguzzi A.
Prions, prionoids and pathogenic proteins in Alzheimer disease.
- Jiang T,
Zhang D,
Chen Q, et al.
TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice.
- Lin T,
Beal F.
Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases.
- Cardoso M,
Santos S,
Swerdlow H,
Oliveira R.
Functional mitochondria are required for amyloid β-mediated neurotoxicity.
- Ghavami S,
Shojaei S,
Yeganeh B, et al.
Autophagy and apoptosis dysfunction in neurodegenerative disorders.
- Chiti F,
Dobson C.
Protein misfolding, functional amyloid, and human disease.
- Reynolds A,
Laurie C,
Moseley RL,
Gendelman HE.
Oxidative stress and the pathogenesis of neurodegenerative disorders.
- Chang S,
ran Ma T,
Miranda D, et al.
Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity.
- Maezawa I,
Maeda N,
Montine J,
Montine KS.
Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice.
- Manzoni C,
Mamais A,
Roosen A, et al.
mTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin-1.
- Hu Q,
Wang G.
Mitochondrial dysfunction in Parkinson’s disease.
- Zhou Q,
Chen B,
Wang X, et al.
Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways.
- Zhu Z,
Yan J,
Jiang W, et al.
Arctigenin effectively ameliorates memory impairment in Alzheimer’s disease model mice targeting both β-amyloid production and clearance.
- Lin K,
Chen D,
Chuang C, et al.
Resveratrol partially prevents rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through induction of heme oxygenase-1 dependent autophagy.
- Peng K,
Tao Y,
Zhang J, et al.
Resveratrol regulates mitochondrial biogenesis and fission/fusion to attenuate rotenone-induced neurotoxicity.
- Csiszár A,
Csiszar A,
Pinto J, et al.
Resveratrol encapsulated in novel fusogenic liposomes activates Nrf2 and attenuates oxidative stress in cerebromicrovascular endothelial cells from aged rats.
- Sarkar S,
Perlstein E,
Imarisio S, et al.
Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models.
- Li W,
Zhu S,
Li J, et al.
EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages.
- Zeng Y,
Zhang J,
Zhu Y,
et al.
Tripchlorolide improves cognitive deficits by reducing amyloid β and upregulating synapse-related proteins in a transgenic model of Alzheimer’s disease.
- Xu P,
Li Z,
Wang H, et al.
Triptolide inhibited cytotoxicity of differentiated PC12 cells induced by amyloid-β25–35 via the autophagy pathway.
- Zhang J,
Wang J,
Xu J, et al.
Curcumin targets the TFEB-lysosome pathway for induction of autophagy.
- Suganthy N,
Devi KP,
Nabavi SF, et al.
Bioactive effects of quercetin in the central nervous system: Focusing on the mechanisms of actions.
- Fukui K,
Masuda A,
Hosono A, et al.
Changes in microtubule-related proteins and autophagy in long-term vitamin E-deficient mice.
- Sasazawa Y,
Sato N,
Umezawa K,
Simizu S.
Conophylline protects cells in cellular models of neurodegenerative diseases by inducing mammalian target of rapamycin (mTOR)-independent autophagy.
- Hsu Y,
Tseng Y,
Lo Y.
Berberine, a natural antidiabetes drug, attenuates glucose neurotoxicity and promotes Nrf2-related neurite outgrowth.
- Rigacci S,
Miceli C,
Nediani C, et al.
Oleuropein aglycone induces autophagy via the AMPK/mTOR signalling pathway: a mechanistic insight.
- Abuznait AH,
Qosa H,
Busnena BA, et al.
Olive-oil-derived oleocanthal enhances β-amyloid clearance as a potential neuroprotective mechanism against Alzheimer’s disease: in vitro and in vivo studies.
- Sheu SJ,
Chen JL,
Bee YS, et al.
Differential autophagic effects of vital dyes in retinal pigment epithelial ARPE-19 and photoreceptor 661W cells.
- Martin L,
Pan Y,
Price A, et al.
Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death.
- Hunn B,
Cragg S,
Bolam J, et al.
Impaired intracellular trafficking defines early Parkinson’s disease.
- Narendra D,
Tanaka A,
Suen DF,
Youle RJ.
Parkin is recruited selectively to impaired mitochondria and promotes their autophagy.
- Lazarou M,
Jin SM,
Kane LA,
Youle RJ.
Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin.
- Zhou ZD,
Sathiyamoorthy S,
Angeles DC,
Tan EK.
Linking F-box protein 7 and Parkin to neuronal degeneration in Parkinson’s disease (PD).
- Jiang J,
Jiang J,
Zuo Y,
Gu Z.
Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson’s disease.
- Le W,
Wu J,
Tang Y.
Protective microglia and their regulation in Parkinson’s disease.
- Wang M,
Cheng X,
Jin M, et al.
TNF compromises lysosome acidification and reduces α-synuclein degradation via autophagy in dopaminergic cells.
- Singh M,
Jensen M,
Lerman A, et al.
Effect of low-dose rapamycin on senescence markers and physical functioning in older adults with coronary artery disease: results of a pilot study.
- Tai H,
Wang Z,
Gong H, et al.
Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence.
- Nikiforov M.
TOR-inhibitors as gero-suppressors.
- Kaeberlein M.
The biology of aging: citizen scientists and their pets as a bridge between research on model organisms and human subjects.
- Deng C,
Tao R,
Yu SZ,
Jin H.
Sulforaphane protects against 6-hydroxydopamine-induced cytotoxicity by increasing expression of heme oxygenase-1 in a PI3K/Akt-dependent manner.
- Jazwa A,
Rojo AI,
Innamorato NG, et al.
Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism.
- Garcia-Alloza M,
Borrelli LA,
Rozkalne A, et al.
Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model.
- Zhang X,
Hu J,
Zhong L, et al.
Quercetin stabilizes apolipoprotein E and reduces brain Aβ levels in amyloid model mice.
- Lee M,
McGeer EG,
McGeer PL.
Quercetin, not caffeine, is a major neuroprotective component in coffee.
- Grimm MO,
Stahlmann CP,
Mett J, et al.
Vitamin E: curse or benefit in Alzheimer’s disease? A systematic investigation of the impact of α-, γ- and δ-tocopherol on Aβ generation and degradation in neuroblastoma cells.
- Chang CF,
Lee YC,
Lee KH, et al.
Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS.
- Zhu ZG,
Sun MX,
Zhang WL, et al. The efficacy and safety of coenzyme Q10 in Parkinson’s disease: a meta-analysis of randomized controlled trials.