Abstract
An epigenetic influence on many chronic and metabolic diseases is now well-established, in some cases with a larger effect on disease risk and susceptibility than many well-established risk factors. Additionally, the evidence base for the role of persistent organic pollutants in many of these same diseases has grown considerably, and it appears that the toxic effects of these substances are mediated in part via epigenetic mechanisms, particularly during critical periods of development. Here we review the associations between these long-lasting toxins and today’s highly prevalent diseases, the epigenetic pathways, as well as suggested interventions to mitigate their harm.
INTRODUCTION
Our understanding of the role of epigenetics in human health and disease has grown considerably in recent years, and is beginning to fill a gap in knowledge not satisfied by sequencing of the genome and genome-wide association studies (GWAS). Although hundreds of genetic polymorphisms which contribute to common traits and diseases have been identified, the cumulative effects of these variants have, at least so far, been surprisingly limited. For example, all GWAS loci combined explain only about 10% of the familial risk for type 2 diabetes. Furthermore, mutations in the genetic sequence do not adequately explain differences in susceptibility to many chronic diseases, including diabetes and obesity, nor how environmental influences communicate with the human genome.
Epigenetics broadly refers to heritable changes in gene function which do not involve variations in DNA sequence. A variety of mechanisms have been studied, including the most widely documented, DNA methylation, as well as alterations in the organization of chromatin within the nucleus, post-translational histone modifications, and regulation by non-coding small RNAs. Chromatin remodeling, for example, alters the accessibility of gene promoters and regulatory regions, thus influencing gene expression. While the permanence of these epigenetic “marks” is still in question, it is clear that most of these changes are quite stable, long-lasting, and likely have trans-generational effects. For instance, a grandparents’ food availability during their “slow growth period” (approximately age 8-12, before a prepubertal peak in growth velocity) was found to be the main determinant of longevity among their grandchildren, giving evidence for a sex-specific transgenerational system, likely mediated via information captured epigenetically. Epigenetic marks may ultimately prove to be as critical to gene and therefore cellular function as is the genetic sequence itself.
Perhaps most importantly, epigenetic modifications appear to be a vital link between gene expression and environmental exposures, including environmental toxicants, nutritional status, and perhaps the social environment. There is now considerable evidence supporting the “Developmental Origins of Adult Health and Disease” (DOHaD) hypothesis, i.e. that during critical windows of development, such as prenatal and early childhood, a lifelong reprogramming of gene expression occurs, setting the stage for disease susceptibility later in life. This is evidenced by the association between both maternal under-nutrition and over-nutrition and a predisposition to develop metabolic syndrome characteristics and cardiovascular disease in offspring. This metabolic plasticity is not necessarily pathological. Rather, epigenetic modifications in general appear to have an adaptive and physiological role, in that they allow for the fetus to make in utero adjustments based upon the predicted postnatal environment. It may instead be the mismatch between the predicted and actual postnatal environment which increases disease susceptibility. Alternatively, and the subject of this paper, exposure to a variety of toxins disrupts this biological communication, interfering with what is likely an evolutionarily adaptive response.
In this review, we discuss the clinical relevance and epigenetic influences of persistent organic pollutants (POPs), long-lasting compounds which bioaccumulate in adipose tissue and have multiple mechanisms of action, including both direct endocrine and oxidative-stress toxicity, as well as more subtle and complex influences on epigenetic programming. We also briefly outline potential therapies and protective interventions.
CLINICAL RELEVANCE OF PERSISTENT ORGANIC POLLUTANTS
The evidence base supporting a link between persistent organic pollutant exposure and a wide spectrum of diseases continues to grow, particularly for cardiometabolic and neurological conditions. These lipophilic and stable substances include a range of substances, such as polychlorinated biphenyls (PCBs), organochlorine pesticides, polybrominated diphenyl ethers (PDBEs), plastics, phthalates, polychlorinated dibenzo-p-dioxins (PCDDs) and polychlorinated dibenzofurans (PCDFs), many of which are widespread globally. While this article is not a comprehensive review of POP disease associations, the following brief summary highlights the clinical importance of these long-lasting substances.
DIABETES, INSULIN RESISTANCE, AND OBESITY:
In a large cross-sectional study of adults, part of the National Health and Nutrition Examination Survey (NHANES) 1999–2002, lipid-adjusted serum levels of six POPs were all found to have a strong positive and dose-dependent correlation with diabetes risk. Indeed, individuals with the highest levels of all six had a 38-fold increase in risk for diabetes compared to those who had serum levels below the limit of detection. Additionally, in those patients with undetectable levels of these six POPs, no association was found between obesity and diabetes, suggesting that excessive adipose tissue may contribute to the risk of diabetes primarily by enhancing the storage and/or toxicity of POPs. Also remarkable is that these six POPs were all chosen not for any particular toxicity, but because they were detectable in >80% of participants.
POP exposure has also been associated with an increase in risk for prediabetes, insulin resistance, and obesity. In a population with heavy POP exposure, a cross-sectional study of over 2000 adults revealed dose-dependent associations between serum levels of five POPs and the risk for both diabetes and prediabetes, with individuals in the upper quintile having a cumulative effect which more than tripled the risk for prediabetes. There also appeared to be an interaction between the five POPs which heightened the diabetes risk more than individual exposure would have predicted.
POPs have been shown to affect several of the root causes of impaired glucose regulation, including the development of insulin resistance, dyslipidemia, inflammation, mitochondrial inhibition, and a decline in pancreatic beta-cell function. Organochlorine (OC) pesticide serum levels were the most strongly associated with homeostasis model assessment of insulin resistance (HOMA-IR) among NHANES participants, with stronger associations at higher waist circumferences, supporting the plausibility of adipose tissue as a mediator of POP toxicity. Further analysis of NHANES data revealed that OC pesticides are also positively associated with C-reactive protein (CRP) levels. Perhaps the most telling finding was that CRP levels were not associated with HOMA-IR among participants with low concentrations of OC pesticides or PCBs, yet CRP levels were strongly associated with HOMA-IR among individuals with high concentrations of OC pesticides or PCBs (Figure 1). These studies question the etiologic importance of inflammation and obesity to insulin resistance, suggesting that POPs may be of greater importance, or at least interact quite significantly with other risk factors. It should be noted that like other endocrine disruptors, POPs do not always display linear dose-dependent effects. Indeed, many display nonmonotonic dose-response curves (NMDRCs), having toxic effects at low doses which are unpredicted by higher dose studies.
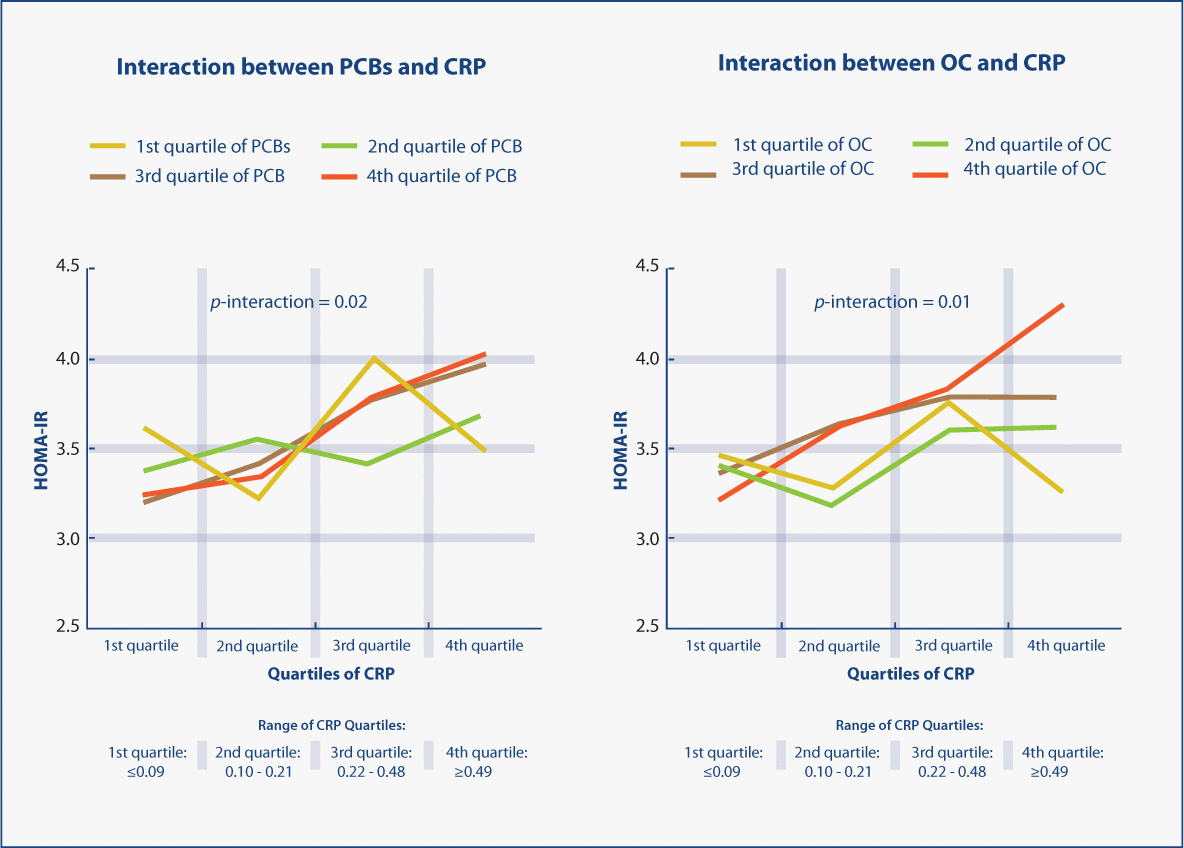
Figure 1: Interaction between C-reactive protein (CRP) and polychlorinated biphenyls (PCBs) or organochlorine (OC) pesticides for estimation of homeostasis model assessment of insulin resistance (HOMA-IR).
Detectable values of each persistent organic pollutant (POP) were individually ranked, and the rank orders of the individual POPs in each subclass were summed to arrive at the subclass value. All undetectable values were ranked as 0. The summary values were categorized by cutoff points of 25th, 50th, and 75th values of the sum of ranks. Geometric means of HOMA-IR values are plotted. Adjusted for age, sex, race, poverty income ratio, body mass index, waist circumference, smoking status, physical activity, alcohol consumption, and OC pesticides (in models including PCBs) or PCBs (in models including OC pesticides).
Kim KS, Hong NS, Jacobs DR Jr, et al. Interaction between persistent organic pollutants and C-reactive protein in estimating insulin resistance among non-diabetic adults. J Prev Med Public Health. 2012 Mar;45(2):62-9. Reprinted under the terms of the Creative Commons Attribution License.
CARDIOVASCULAR, DEVELOPMENTAL, AND NEUROLOGICAL TOXICITY:
Data from NHANES also implicates POP exposure as a risk factor for cardiovascular disease. Self-reported cardiovascular disease and hypertension have both been associated with higher serum levels of specific POPs, including both dioxin-like PCBs and non-dioxin-like PCBs. Differing effects on gender were seen for specific classes of POPs; for example, PCDDs had as much as a 5-fold increase in risk for hypertension among women in the highest quartile, with no association in men, while PCBs had a 2-3 fold increase in risk among men, but not among women. In a more recent analysis, higher serum levels of OC pesticides were found to be a potent risk factor for peripheral arterial disease, but only among obese participants. In two cross-sectional studies from the populaton-based Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS), serum POP levels were found to be predictive of carotid atherosclerosis. PCBs, phthalates, and bisphenol A (BPA) were found to be associated with plaque formation as well as the echogenicity of the intima-media complex, a marker for a lipid-rich vascular wall.
POP exposure has also been associated with developmental toxicity and cognitive impairment. One in a series of three studies published in Environmental Health Perspectives, an analysis of a birth cohort in an agricultural area revealed an association between urinary metabolites of organophosphate (OP) pesticides during pregnancy and impaired intellectual development among children at age seven. Those children whose mothers were in the highest quintile for exposure had an average 7 point reduction in IQ compared to those in the lowest. While the children’s own urinary levels were not associated with intellectual development, 25% of women in the US have urinary levels exceeding the median of mothers in this study, suggesting potentially widespread harm. A second study had similar findings, reporting higher prenatal exposure to chlorpyrifos (CPF) to be associated with both a lower IQ as well as lower “working memory”, a characteristic tied to attention capacity. This is in agreement with previous findings that higher urinary levels of OP pesticide metabolites among children increases the risk for attention deficit disorder, and here too, the levels associated with harm are relatively common in the U.S. A number of structural changes in the brains of children who had high prenatal CPF exposure have also been documented. The third study in this series not only found prenatal OP pesticide urinary levels to be associated with impaired cognitive development among children, but variants in the maternal PON1 gene, which is involved in OP pesticide metabolism, modulated this effect. Lastly, in addition to OP pesticides, other POPs have also demonstrated developmental effects. Prenatal and postnatal exposure to polybrominated diphenyl ethers, for example, has been associated with poor social competence and attention deficit in children, as well as lower IQ and other physical and cognitive developmental scores. Phthalate and polyfluoroalkyl chemicals (PFCs) are also implicated.
In addition to developmental impairment, POP exposure has also been associated with cognitive impairment and neurodegenerative disease risk. For example, variants in the gene mentioned above, PON1, more than doubled the risk for Parkinson’s disease among exposed individuals. In a recently published meta-analysis, insecticide and herbicide exposure had a summary risk ratio (sRR) of 1.62 for Parkinson’s disease, while exposure based upon job-title had a 2.5 sRR. Pesticide exposure, particularly OC pesticides, has also been associated with an increase in risk for amyotrophic lateral sclerosis as well.
EPIGENETIC INFLUENCE OF POPS
POPS AND HYPOMETHYLATION:
Among the first studies to document alterations in DNA methylation associated with exposure to POPs was conducted among the Greenland Inuit, a population with the highest concentration of POPs measured in humans worldwide. Using percent 5-methylcytosine (%5-mC), a surrogate marker for percentage of global genomic DNA methylation, researchers cited a consistent inverse correlation and linear relationship between POP levels and %5-mC. Given its cross-sectional design, cause and effect could not be determined, but a strong association between POP burden and global hypomethylation is quite significant. Global DNA hypomethylation is frequently associated with chromosomal instability and expression of genes normally silenced by methylation, and is a nearly universal finding among cancer tissues and cancer precursor cells.
A second study conducted among apparently healthy Korean adults also found that a higher POP burden (quantified by lipid-adjusted serum levels) was also associated with reduced %5-mC assays, with OC pesticides having the strongest and most consistent association. This study is particularly relevant because the POP burden in this population is not at the extremes, such as in the Greenland Inuit. Rather, it is quite comparable to the U.S. general population burden. Additionally the authors point out that the association between POPs and hypomethylation was similar in magnitude to the correlation between age and global hypomethylation. Adjusting for age did not affect the results, suggesting POP accumulation over the lifespan may partly explain the well-known association between aging and global hypomethylation.
CANCER RISK AND METHYLATION:
We now have evidence that methylation status may be a screening tool for cancer risk among humans. Friso et al. established that the peripheral blood mononuclear cell (PBMCs) DNA methylation status of individuals with cancer was “invariably decreased compared with control group subjects, indicating that cancer subjects have a systemically decreased global DNA methylation”. They also found that global DNA methylation in PBMCs was lower in the individuals who developed cancer during the eight years of follow-up compared to cancer-free controls. Additionally, decreased DNA methylation status related with lower folate status and higher frequency of the MTHFR 677TT genotype, and the latter increased cancer risk only in those who also had low folate status (OR for cancer of 7.042, CIs 1.52–32.63, P=0.013). This both points the way toward the use of methylation as a screening tool for disease susceptibility, and highlights the interaction of diet, environment, epigenome, and genome in determining risk. Similarly, Godderis et al. reported that solvent exposure as well as polymorphisms in the glutathione S-transferase P gene were associated with global hypomethylation. It has been proposed that POP exposure leads to hypomethylation because the methylation cycle and the glutathione synthesis pathway are linked (a “unifying hypothesis”). Thus additional need for glutathione, which is the main route of detoxification for many POPs, depletes the supply of other methyl donors, such as S-adenosylmethionine.
DIABETES, OBESITY & METHYLATION:
Hypomethylation also appears to have a sizeable impact on diabetes risk. A genome-scale screen among nearly 1200 individuals found differentially methylated sites in genomic regions that were previously associated with diabetes risk, establishing a diabetes methylation pattern. They determined that the methylation level at just a single site (the FTO methylation site) “is more closely related to T2DM (area under the curve (AUC) = 0.638, 95% CI: 0.586–0.690) than the sequence variant with the largest effect identified to date (rs7901695 in the TCF7L2 LD block, AUC = 0.55), or the 18 most established genetic variants combined (AUC = 0.6)”, quite a remarkable finding. Specifically, the odds of belonging to the diabetes group increased by 6.1% for every 1% decrease in methylation, with hypomethylation increasing the risk of diabetes in both genders. The authors were surprised to find no correlation between methylation and BMI, with cases hypomethylated relative to controls in both obese and non-obese subjects, suggesting “that the observed association with T2DM is not mediated through obesity”, despite a well-established association between FTO alleles and obesity. Additionally, they documented that among 515 initially healthy participants, those who progressed to impaired glucose homeostasis were hypomethylated relative to controls (P = 0.019), indicating that “low methylation level is not the result of the disease, but rather an early risk factor that predisposes to disease manifestation later in life.” It is important to note that risk for diabetes may be modified by differential methylation at specific sites rather than an absolute increase or decrease in methylation. For example, hypermethylation of the insulin promoter has been found in patients with type 2 diabetes, negatively correlating with insulin gene expression in human pancreatic islets.
Methylation status also appears quite relevant to the risk of obesity. Increased methylation of RXRA chr9:136355885+ (extracted from umbilical cord tissue at birth) was found to be associated with a greater degree of adiposity in later childhood, in two independent cohorts. Indeed, this study suggested that “a substantial proportion of the variation in adiposity in prepubertal children can be explained by epigenetic measurements made at birth”, with an effect “considerably greater than that of factors such as birth weight or maternal body composition” (Figure 2) Notably, POPs such as phthalates have been associated with body size and fat distribution in both children and the elderly, as well as insulin resistance and diabetes prevalence.
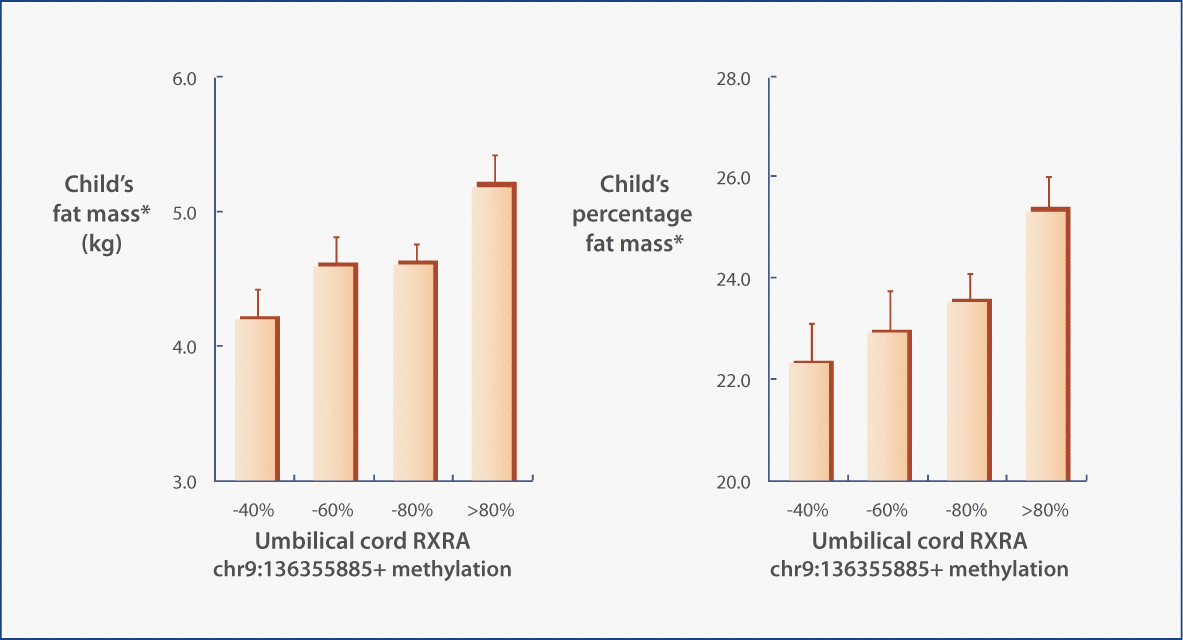
Figure 2: In a second independent cohort, child’s % fat mass and fat mass at age 6 years increase with higher umbilical cord RXRA chr9:136355885+ methylation in SWS subjects. Values are means + SEM. *Fat mass and percentage fat mass are preadjusted for age and sex.
Godfrey KM, Sheppard A, Gluckman PD, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011 May;60(5):1528-34. Reprinted under the terms of the Creative Commons Attribution License.
ADDITIONAL EPIGENETIC INFLUENCES OF POPS
Though not a comprehensive review, it is important to note that the influence of POPs on epigenetic markers of disease extends well beyond cancer, diabetes, and obesity. Hypomethylation and altered methylation patterns have been documented among patients with heart failure and in atherosclerotic plaques. Although an increase in plasma homocysteine is positively associated with atherosclerosis and cardiovascular disease risk, the true risk may be from impaired methylation capacity for which homocysteinemia is the end result. Thus it may be that S-adenosylhomocysteine and/or S-adenosylmethionine are better biomarkers of risk.
As mentioned above, pesticide exposure, particularly dieldrin, has been associated with an increase in risk for Parkinson’s disease. This may be in part due to an epigenetic influence, as recently dieldrin has been found to rapidly induce the hyperacetylation of histones in dopaminergic neuronal cells, “an early signaling event in the execution of apoptosis”. Bisphenol A is one of the most well-documented endocrine disruptors, yet it appears that some of its adverse effects “on brain development, sexual differentiation and behavior, as well as immune function” may be mediated by epigenetic modulation, including alterations to DNA methylation patterns. Prenatal exposure has been associated with asthma, abnormal behaviors in children, including hyperactivity, aggression, anxiety, and depression, as well as adverse birth outcomes. Autism spectrum disorder has been associated with impaired glutathione availability, reduced detoxification capacity, and hypomethylation, of which the latter is also found in parents of autistic children, and both PCBs and PBDE congeners appear to influence risk. Methoxychlor, a persistent and bioacummulating organochlorine insecticide, has shown in animal studies to cause lifelong reprogramming in neuroendocrine gene expression and DNA methylation, which result in the advancement of reproductive senescence. Asthma, neurodegenerative, inflammatory, and reproductive disorders, as well as the aging process itself appear to be influenced by POP mediated epigenetic changes. Lastly, in addition to modifications to nuclear DNA, mitochondrial DNA has its own epigenetic programming, potentially an additional mechanism by which POPs (which are known to impair mitochondrial function) exert lasting toxicity.
SUGGESTED INTERVENTIONS:
Avoidance of POP exposure is primarily accomplished through dietary means. Because these toxins bioaccumulate in adipose tissue and up the food chain, an organic and largely plant-based diet may be the most effective for reducing exposure. For example, diet has been shown to be the largest source of exposure to OP pesticides in young children, and an organic diet dramatically reduces urinary metabolites levels. Diet is not the only source of exposure to many POPs; phthalates are found in many personal care products, and BPA in many food containers. Exercise, particularly more vigorous exercise, has also shown an effect on epigenetic markers such as DNA methylation, and may mediate benefit via several mechanisms.
As we have reviewed elsewhere, in addition to serum and urinary markers of exposure to specific pollutants, serum GGT may be a reasonable and inexpensive biomarker for POP exposure, as it represents an increased need for GSH synthesis. Thus providing support for glutathione synthesis either directly or with precursors such as N-acetylcysteine may be indicated in those at greater risk or exposure levels. Lastly, providing adequate methyl donors, such as folic acid, betaine, B12, etc., appears to reduce the risk of conditions ranging from autism to cardiovascular disease, at least in part via epigenetic mechanisms, and may be indicated in those with high exposure and/or signs of impaired methylation capacity.
CONCLUSION
Persistent organic pollutants have emerged as important contributors to many of today’s chronic diseases, perhaps more so than many well-accepted risk factors. POPs display synergistic and low-dose effects that have allowed them to escape the closer regulatory scrutiny which is given to high dose and directly mutagenic toxins. At least part of their toxicity appears to be mediated via epigenetic mechanisms, which now appear to be on par with genetic polymorphisms for clinical importance. Unlike genetic mutations, epigenetic modifications appear to be reversible with appropriate interventions. While much remains to be determined, avoidance of exposure seems highly indicated, as does nutritional support of relevant metabolic pathways, including glutathione synthesis and methylation, particularly during crucial periods of development.
DISCLOSURE OF INTERESTS
Dr. J.E. Pizzorno reports personal fees from Bioclinic Naturals for dietary supplements, outside the submitted work. Dr. Katzinger has nothing to disclose.
REFERENCES
- Maher B. Personal genomes: The case of the missing heritability.
- Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS?
- Kussmann M, Krause L, Siffert W. Nutrigenomics: where are we with genetic and epigenetic markers for disposition and susceptibility?
- Kaati G, Bygren LO, Pembrey M, et al. Transgenerational response to nutrition, early life circumstances and longevity.
- Pembrey ME. Male-line transgenerational responses in humans.
- Szyf M, McGowan P, Meaney MJ. The social environment and the epigenome.
- Armitage JA, Taylor PD, Poston L. Experimental models of developmental programming: consequences of exposure to an energy rich diet during development.
- Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective.
- Porta M, Puigdomenech E, Ballester F, et al. Monitoring concentrations of persistent organic pollutants in the general population: the international experience.
- Lee DH, Lee IK, Song K, et al. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999-2002.
- Ukropec J, Radikova Z, Huckova M, et al. High prevalence of prediabetes and diabetes in a population exposed to high levels of an organochlorine cocktail.
- Lee DH, Steffes MW, Sjödin A, et al. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes.
- Lim S, Ahn SY, Song IC, et al. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance.
- Lee HK. Mitochondrial dysfunction and insulin resistance: the contribution of dioxin-like substances.
- Lee DH, Lee IK, Jin SH, et al. Association between serum concentrations of persistent organic pollutants and insulin resistance among nondiabetic adults: results from the National Health and Nutrition Examination Survey 1999–2002.
- Kim KS, Hong NS, Jacobs DR Jret al. Interaction Between Persistent Organic Pollutants and C-reactive Protein in Estimating Insulin Resistance Among Non-diabetic Adults.
- Vandenberg LN, Colborn T, Hayes TB, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses.
- Ha M.H, Lee D.H, et al. Association between serum concentrations of persistent organic pollutants and self-reported cardiovascular disease prevalence: results from the National Health and Nutrition Examination Survey, 1999–2002.
- Ha M.H, Lee D.H, et al. Association between serum concentrations of persistent organic pollutants and prevalence of newly diagnosed hypertension: results from the National Health and Nutrition Examination Survey 1999–2002.
- Min JY, Cho JS, Lee KJ, et al. Potential role for organochlorine pesticides in the prevalence of peripheral arterial diseases in obese persons: results from the National Health and Nutrition Examination Survey 1999–2004.
- Lind PM, van Bavel B, Salihovic S, et al. Circulating levels of persistent organic pollutants (POPs) and carotid atherosclerosis in the elderly.
- Lind PM, Lind L. Circulating levels of bisphenol A and phthalates are related to carotid atherosclerosis in the elderly.
- Bouchard MF, Chevrier J, Harley KG, et al. Prenatal exposure to organophosphate pesticides and IQ in 7-year-old children.
- Rauh V, Arunajadai S, Horton M, et al. Seven-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide.
- Bouchard MF, Bellinger DC, Wright RO, et al. Attention-deficit/hyperactivity disorder and urinary metabolites of organophosphate pesticides.
- Rauh VA, Perera FP, Horton MK, et al. Brain anomalies in children exposed prenatally to a common organophosphate pesticide.
- Engel SM, Wetmur J, Chen J, et al. Prenatal exposure to organophosphates, paraoxonase 1, and cognitive development in childhood.
- Gascon M, Vrijheid M, Martínez D, et al. Effects of pre and postnatal exposure to low levels of polybromodiphenyl ethers on neurodevelopment and thyroid hormone levels at 4 years of age.
- Herbstman JB, Sjödin A, Kurzon M, et al. Prenatal exposure to PBDEs and neurodevelopment.
- Eskenazi B, Chevrier J, Rauch SA, et al. In utero and childhood polybrominated diphenyl ether (PBDE) exposures and neurodevelopment in the CHAMACOS Study.
- Hoffman K, Webster TF, Weisskopf MG, et al. Exposure to polyfluoroalkyl chemicals and attention deficit/hyperactivity disorder in U.S. children 12–15 years of age.
- Engel SM, Miodovnik A, Canfield RL, et al. Prenatal phthalate exposure is associated with childhood behavior and executive functioning.
- Whyatt RM, Liu X, Rauh VA, et al. Maternal prenatal urinary phthalate metabolite concentrations and child mental, psychomotor, and behavioral development at 3 years of age.
- Manthripragada AD, Costello S, Cockburn MG, et al. Paraoxonase 1, agricultural organophosphate exposure, and Parkinson disease.
- van der Mark M, Brouwer M, Kromhout H, et al. Is pesticide use related to Parkinson disease? Some clues to heterogeneity in study results.
- Malek AM, Barchowsky A, Bowser R, et al. Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: a meta-analysis of epidemiological studies: pesticide exposure as a risk factor for ALS.
- Kamel F, Umbach DM, Bedlack RS, et al. Pesticide exposure and amyotrophic lateral sclerosis.
- Dewailly E, Mulvad G, Pedersen HS, et al. Concentration of organochlorines in human brain, liver, and adipose tissue autopsy samples from Greenland.
- Rusiecki JA, Baccarelli A, Bollati V, et al. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit.
- Ehrlich M. Cancer-linked DNA hypomethylation and its relationship to hypermethylation.
- Goelz SE, Vogelstein B, Hamilton SR, et al. Hypomethylation of DNA from benign and malignant human colon neoplasms.
- Kim KY, Kim DS, Lee SK, et al. Association of low-dose exposure to persistent organic pollutants with global DNA hypomethylation in healthy Koreans.
- Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases.
- Friso S, Udali S, Guarini P, et al. Global DNA hypomethylation in peripheral blood mononuclear cells as a biomarker of cancer risk.
- Godderis L, De Raedt K, Tabish AM, et al. Epigenetic changes in lymphocytes of solvent-exposed individuals.
- Lee DH, Jacobs DR Jr, Porta M. Hypothesis: a unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation.
- Toperoff G, Aran D, Kark JD, et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood.
- Yang BT, Dayeh TA, Volkov PA, et al. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes.
- Godfrey KM, Sheppard A, Gluckman PD, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity.
- Lind PM, Roos V, Rönn M, et al. Serum concentrations of phthalate metabolites related to abdominal fat distribution two years later in elderly women.
- Teitelbaum SL, Mervish N, Moshier EL, et al. Associations between phthalate metabolite urinary concentrations and body size measures in New York City children.
- Lind PM, Zethelius B, Lind L. Circulating Levels of Phthalate Metabolites Are Associated With Prevalent Diabetes in the Elderly.
- Movassagh M, Vujic A, Foo R. Genome-wide DNA methylation in human heart failure.
- Wagner C, Koury MJ. S-Adenosylhomocysteine: a better indicator of vascular disease than homocysteine?
- Baccarelli A, Ghosh S. Environmental exposures, epigenetics and cardiovascular disease.
- Castro R, Rivera I, Struys EA, et al. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease.
- Song C, Kanthasamy A, Anantharam V, et al. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration.
- Kundakovic M, Champagne FA. Epigenetic perspective on the developmental effects of bisphenol A.
- Wolstenholme JT, Rissman EF, Connelly JJ. The role of Bisphenol A in shaping the brain, epigenome and behavior.
- Spanier AJ, Kahn RS, Kunselman AR, et al. Prenatal exposure to bisphenol a and child wheeze from birth to 3 years of age.
- Braun JM, Kalkbrenner AE, Calafat AM, et al. Impact of early-life bisphenol A exposure on behavior and executive function in children.
- Chou WC, Chen JL, Lin CF, et al. Biomonitoring of bisphenol A concentrations in maternal and umbilical cord blood in regard to birth outcomes and adipokine expression: a birth cohort study in Taiwan.
- James SJ, Melnyk S, Jernigan S, et al. Abnormal transmethylation/transsulfuration metabolism and DNA hypomethylation among parents of children with autism.
- Melnyk S, Fuchs GJ, Schulz Eet al. Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism.
- Mitchell MM, Woods R, Chi LHet al. Levels of select PCB and PBDE congeners in human postmortem brain reveal possible environmental involvement in 15q11-q13 duplication autism spectrum disorder.
- Woods R, Vallero RO, Golub MS, et al. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation.
- Gore AC, Walker DM, Zama AM, et al. Early life exposure to endocrine-disrupting chemicals causes lifelong molecular reprogramming of the hypothalamus and premature reproductive aging.
- Cortessis VK, Thomas DC, Levine AJ, et al. Environmental epigenetics: prospects for studying epigenetic mediation of exposure-response relationships.
- Salam MT, Byun HM, Lurmann F, et al. Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children.
- Wallace DC. Bioenergetics and the epigenome: interface between the environment and genes in common diseases.
- Curl CL, Fenske RA, Elgethun K. Organophosphorus pesticide exposure of urban and suburban preschool children with organic and conventional diets.
- Lu C, Barr DB, Pearson MA, et al. Dietary intake and its contribution to longitudinal organophosphorus pesticide exposure in urban/suburban children.
- Barrès R, Yan J, Egan Bet al. Acute exercise remodels promoter methylation in human skeletal muscle.
- Pizzorno, J, Katzinger, J. Glutathione: Physiological and Clinical Relevance.
- LaSalle JM. A genomic point-of-view on environmental factors influencing the human brain methylome.
- Baccarelli A, Cassano PA, Litonjua Aet al. Cardiac autonomic dysfunction: effects from particulate air pollution and protection by dietary methyl nutrients and metabolic polymorphisms.