Abstract
We have reached a limit in our ability to reduce the incidence of coronary heart disease and cardiovascular disease utilizing the traditional evaluation, prevention, and treatment strategies for the top five cardiovascular risk factors—hypertension, diabetes mellitus, dyslipidemia, obesity and smoking. Statistics show that approximately 50% of patients continue to have coronary heart disease or myocardial infarction despite traditionally defined “normal” levels of these five risk factors. A more logical and in-depth understanding is required of these top five risk factors, including, the evaluation of 24-hour ambulatory blood pressure monitoring, advanced lipid profiles, dysglycemic parameters, visceral obesity, with the effects of adipokines and of the three finite vascular endothelial responses (which include inflammation, oxidative stress and immune vascular dysfunction) to the infinite number of insults. Understanding translational cardiovascular medicine to correlate the coronary heart disease risk factors to the presence or absence of vascular injury and disease with non-invasive vascular testing will allow for early identification, prevention and treatment of coronary heart disease and cardiovascular disease.
INTRODUCTION
Cardiovascular medicine needs a complete functional and metabolic reevaluation related to diagnosis, prevention, and integrative treatments. We have reached a limit in our ability to treat cardiovascular disease (CVD) appropriately. Cardiovascular disease is a group of vascular and heart disorders which include coronary heart disease, cerebrovascular disease, peripheral arterial disease, rheumatic heart disease, congenital heart disease and deep vein thrombosis and pulmonary embolism. In these states the cardiovascular system is literarily “on fire”; our present treatments are not effective at reducing this vascular inflammation. CVD remains the number one cause of morbidity and mortality in the United States.$80 billion a year is spent treating CVD alone in the United States and over 2200 US citizens die from stroke or myocardial infarction (MI) each day. Coronary heart disease (CHD) includes angina, MI, ischemic heart disease, ischemic cardiomyopathy with both systolic (low ejection fraction) and diastolic congestive heart failure (CHF; normal ejection fraction with stiff and non-compliant left ventricle). Ischemic heart disease is the leading cause of death in the world responsible for approximately 17 million deaths in 2011. Of these, 41% were due to ischemic heart disease and 36% due to stroke
The traditional evaluation, prevention, and treatment strategies for the top five cardiovascular risk factors—hypertension, diabetes mellitus, dyslipidemia, obesity, and smoking, have resulted in what is now referred to as a “CHD gap”. Approximately 50% of patients continue to have CHD or MI despite having “normal” levels of these risk factors, as currently defined in the medical literature. We maintain a cholesterol-centric approach to the management of CHD, but do not address the basic etiologies of CHD such as inflammation, oxidative stress and immune vascular dysfunction. However, there are important details within each of these top five risk factors that are not being measured by physicians and are thus ignored in the prevention and treatment of CHD. In fact, there are at least 395 other risk factors that physicians either do not know about, or they are not using appropriate techniques to identify and treat them. Thus, it is imperative that we now begin to examine other methods to prevent and treat CVD.
REVOLUTIONIZING THE TREATMENT OF CARDIOVASCULAR DISEASE
The blood vessel has three known finite responses to an infinite number of insults. Those responses are inflammation (the body’s response to attempt to remove the injurous stimuli and initiate healing), oxidative stress (an imbalance between reactive oxygen species and the body’s ability to detoxify the reactive intermediates), and vascular immune dysfunction (related to T cell and B cell function). Tracking backwards from those three finite responses brings us to the genesis of CVD, with the goal of starting effective treatments to resolve the downstream abnormalities.
Cell membrane physiology and cell membrane dysfunction are keys to this treatment strategy. The membrane barrier between the outside and the inside of every one of our cells, such as the endothelium, enterocyte, the blood–brain barrier, or any other membrane, regulates many of the signaling mechanisms that occur from the external to the internal milieu.
Any cell membrane insult such as high blood pressure (BP), low density lipoprotein (LDL) cholesterol, glucose, microbes, toxins, heavy metals or homocysteine results in a reaction diffusion wave throughout the cell membrane that disrupts the signaling mechanisms and induces membrane damage and dysfunction. The reaction diffusion wave means that a single injury point will be transmitted throughout the cell membrane to induce injury. One small insult becomes a heightened response (metabolic memory) to create further cell damage. This reaction to a foreign insult is an appropriate response by the body, but the blood vessel is injured in a cross reaction, and is really an innocent bystander in a correct but often dysregulated vascular response to these infinite insults.
In the acute setting, any vascular insult results in a correct defensive response by the endothelium. The vascular immune dysfunction, oxidative stress or inflammatory responses are usually short-lived, appropriate, and regulated. However, chronic insults result in a chronic exaggerated and dysregulated vascular dysfunction with preclinical then clinical CVD due to maladaptation of various systems such as the renin-angiotensin-aldosterone (RAAS) system, sympathetic nervous system (SNS) and others.
Most diseases are defined with a specific abnormal level. Hypertension is defined as systolic blood pressure >140 and/or diastolic blood pressure >90 mmHg, dyslipidemia as an LDL-cholesterol of >100 mg/dl, and glucose intolerance as a fasting glucose level of >99 mg/dl. However, it is very clear that there is a continuum of risk starting at lower levels of BP, LDL cholesterol, and glucose, as well as for most other CHD risk factors. For example, we know that the BP risk for CVD actually starts at 110/70 mmHg, and that the risk of LDL cholesterol causing reduction in nitric oxide in the endothelium starts at 60 mg/dl and fasting glucose risk starts at 75 mg/dl. There is a progressive continuum of risk with all of the CVD risk factors and mediators that affect the blood vessel, leading initially to functional abnormalities (endothelial dysfunction), then to structural abnormalities of the vascular and cardiac muscle and to preclinical and clinical CVD. In the case of LDL cholesterol, the increase in risk is non-linear, rising more steeply with increasing LDL-C concentrations. Blood pressure also follows a non-linear model, while glucose follows a threshold model with linear slope and blood pressure.
It is important to understand the concept of “translational vascular medicine.” For example: do the risk factors that are measured actually translate into a vascular illness? Then, vice versa: does the absence of those risk factors actually define vascular health? At this time we often do not use functional and structural markers of vascular and endothelial dysfunction to identify the vascular effects of CHD risk factors or the presence of vascular disease. Instead, we are relying only upon risk factors or some risk factor scoring system such as Framingham or COSEHC (Consortium of Southeastern Hypertension Centers). We assume that if a patient has risk factors, they also have vascular disease; but if they don’t have risk factors, they may have vascular health. It is important to measure sensitive indicators of endothelial dysfunction and vascular structural disease that are induced by the insults for early detection of risk for cardiovascular events. Early detection with aggressive treatment may reduce CVD.
THE ENDOTHELIUM, ENDOTHELIAL FUNCTION, AND ENDOTHELIAL DYSFUNCTION
The endothelium is a very thin lining of vascular cells which forms an interface between the circulating blood in the lumen and the vascular smooth muscle. When the endothelium is working correctly (endothelial function) all the blood elements and the vascular smooth muscle remain normal. However, when endothelial dysfunction occurs, the results are inflammation, oxidative stress, immune dysfunction, abnormal growth, vasoconstriction, increased permeability, thrombosis and ultimately CVD.
Figure 1 illustrates the role of LDL cholesterol in atherosclerotic plaque formation. Once inside the vessel wall, LDL cholesterol becomes susceptible to oxidation and modification by free radicals and glycation. Oxidized LDL and glycated LDL are toxic to the vessel wall. The modified LDL is consumed by scavenger receptors (SR-A and CD-36) on macrophages to form foam cells. Foam cells are not able to process the oxidized LDL or modified LDL and continue to accumulate oxidized and modified LDL, forming a plaque which may rupture and cause acute coronary thrombosis. This is the progression that needs to be interrupted. There are actually 38 different steps in this process that are important in the treatment of dyslipidemia-induced vascular disease (e.g. glucose, lipids, homocysteine, heavy metals) that are promising therapeutic targets (e.g. with ARBs, exercise, statins).
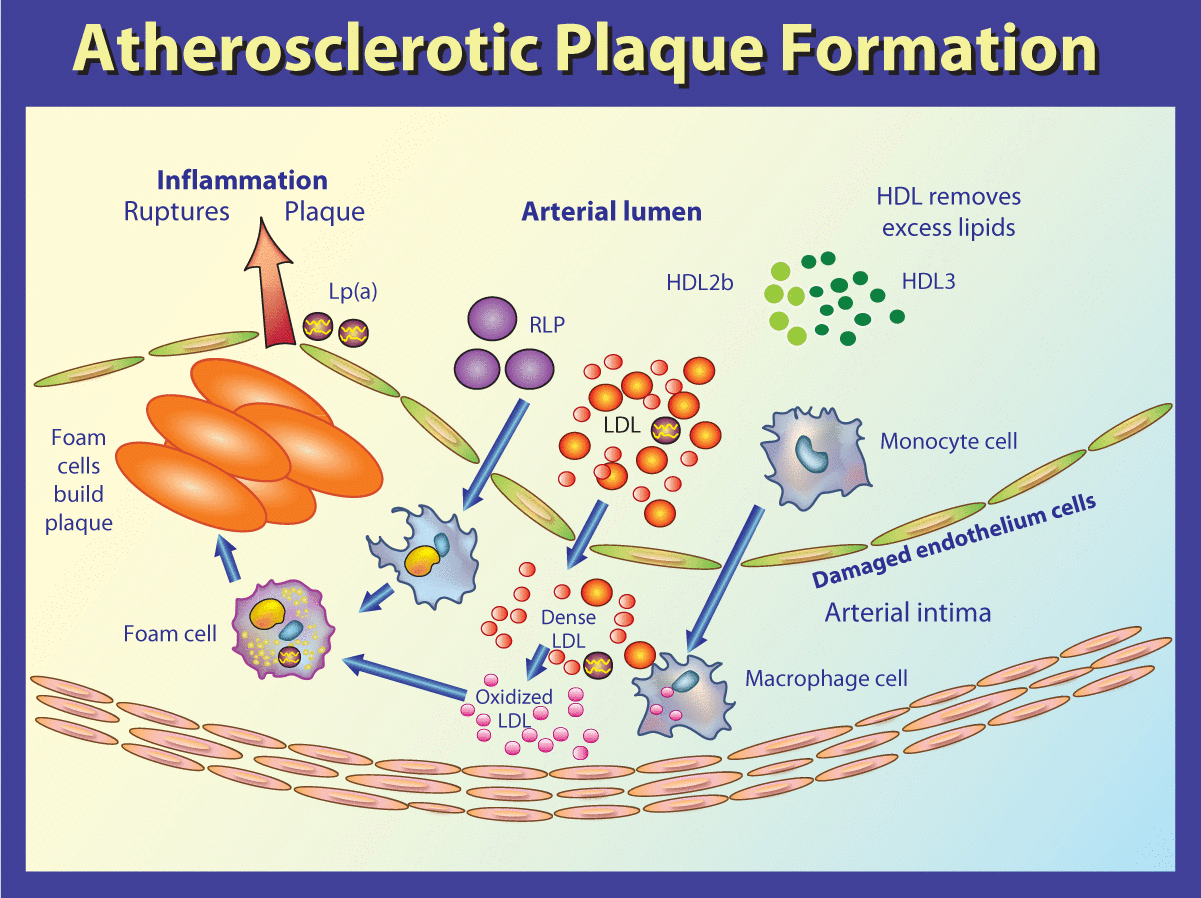
A simplified illustration of atherosclerotic plaque formation.
Vascular disease is a balance of vascular injury (induced by contracting factors such as angiotensin II and endothelin) versus vascular repair with endothelial progenitor cells (EPCs) produced in the bone marrow. The infinite insults result in preconditioned and heightened “metabolic memory” responses that trigger the three known finite downstream responses that have a bi-directional communication involving endothelial dysfunction, vascular smooth muscle, and cardiac dysfunction. Once endothelial dysfunction has developed, a smaller insult occurring at a later stage can result in a heightened response that induces more vascular damage. The concept of metabolic memory was demonstrated by Youssef-Elabd et al. who found that short-term exposure of adipose cells to uncontrolled levels of saturated fatty acids and glucose led to a long-term inflammatory insult within adipocytes.
THE PATHOPHYSIOLOGY OF VASCULAR DISEASE
The major causes of vascular disease are:
Oxidative stress:
Oxidative stress:25,26 reactive oxygen species (ROS) and reactive nitrogen species (RNS) are increased in the arteries and kidneys and with decreased oxidative defenses.
- Inflammation: this is increased in the vasculature and kidneys and includes increased C-reactive protein (CRP), leukocytosis, increased neutrophils and decreased lymphocytes, and upregulation of RAAS in the kidney.
- Autoimmune dysfunction: of the arteries and kidneys, which includes, increased white blood count (WBC), and involvement of CD4+ (T-helper cells) and CD8+ (cytotoxic T-cells).
These problems result in abnormal vascular biology with endothelial dysfunction and vascular smooth muscle hypertrophy and dysfunction. Of course, genetics, genomics, and epigenetics also play a role in the pathophysiology of vascular disease.
Figure 2 offers an insight into the infinite insults that the endothelium is bombarded by. The infinite insults are divided into two major categories: biomechanical (BP, pulse pressure, shear stress, and oscillatory pressure within the arterial system—most plaques form at the bifurcation of arteries), and biochemical (e.g., nutritional factors, microbes, sterile antigens, and environmental toxins).
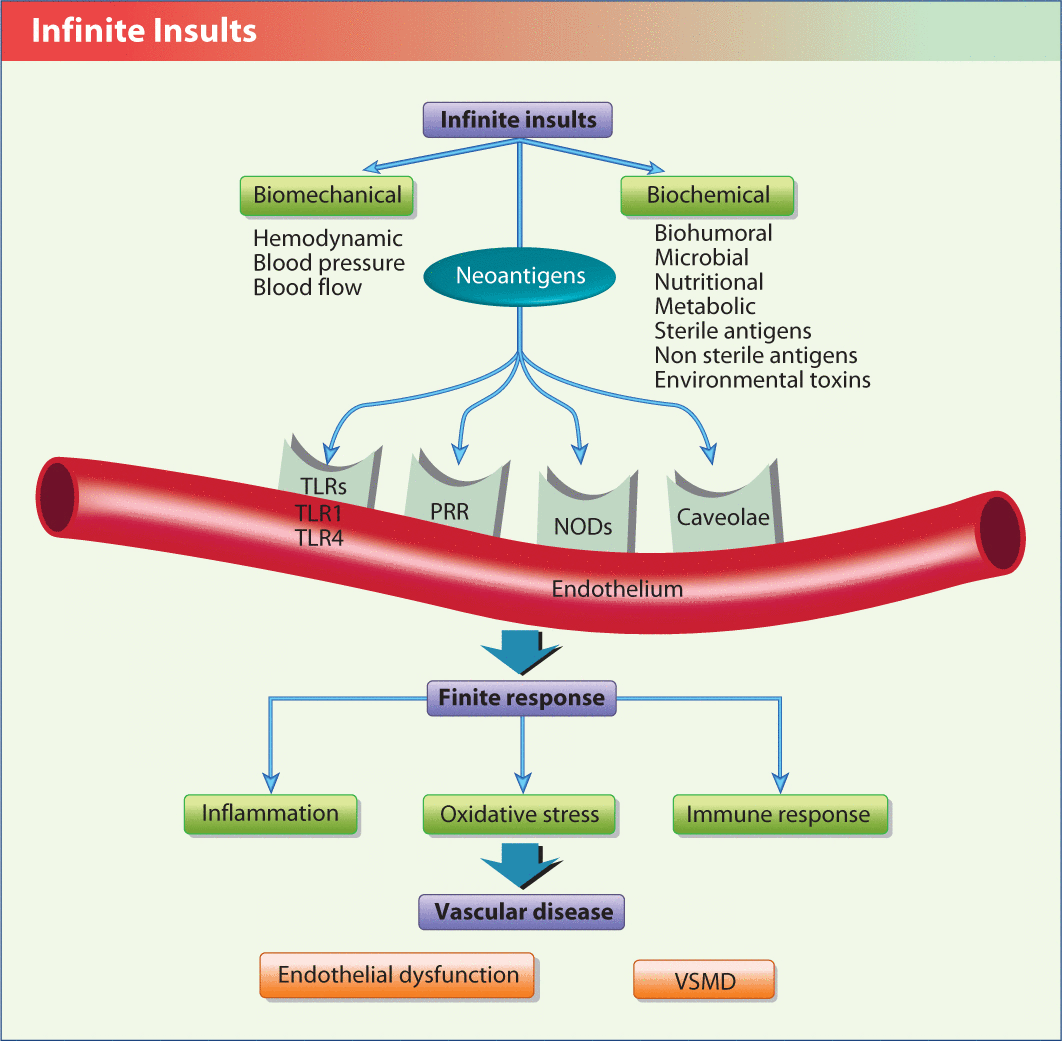
The endothelium is subject to an infinite number of insults but can only elicit a finite number of responses to those insults.
NOD, nucleotide-binding oligomerization domain receptor; PRR, pattern recognition receptor; TLR, toll-like receptor; VSMD,
Endothelial cells express various receptors that determine the interaction between the insults and the downstream mediators. These include pattern recognition receptors (PRR), toll-like receptors (TLR), nod-like receptors (NLR), and caveolae. The TLRs and NLRs are membrane receptors that react to external insults with appropriate intracellular signaling that usually induces inflammation, oxidative stress and immune dysfunction within the cell. The caveolae are membrane lipid microdomains that, when interrupted or stimulated, reduce nitric oxide levels, and increase BP, inflammation, dyslipidemia, oxidative stress and immune dysfunction leading to atherosclerosis. The various risk factors and risk mediators attach to one of the receptors in the membrane and then set off a cascade of the three finite responses (inflammation, oxidative stress, and immune dysfunction), which leads to endothelial dysfunction and ultimately CVD.
INTERRUPTING THE FINITE PATHWAYS
The key to the successful prevention and treatment of CVD is recognition of risk factors, modifying risk factors and interrupting pathways that connect the risk factors to the receptors discussed above. The TLR 1, 2 and 4 are the most common of the PRR type TLRs related to the vascular membrane and endothelial dysfunction. TLR 2 and TLR4 are most important, but TLR2 activation may occur via a TLR1 or TLR-6 coreceptor independent pathway. The NLRs: NOD 1 and NOD 2 are also types of PRRs that involve the vascular membrane. There are many scientifically proven nutraceuticals and dietary factors that accomplish this:
- Curcurmin (turmeric): the PRRs, TLR 4, NOD 1 (NLR), and NOD 2 (NLR)
- Cinnamaldehyde (cinnamon): TLR 4
- Sulforaphane (broccoli): TLR 4
- Resveratrol (nutritional supplement, red wine, grapes): TLR 4
- Epigallocatechin gallate (green tea): TLR 4
- Luteolin (celery, green pepper, rosemary, carrots, oregano, oranges, olives): TLR 4
- Quercetin (tea, apples, onion, tomatoes, capers): TLR 4
- Chrysin: TLR 1
- Omega 3 fatty acids: Interrupt caveolae lipid microdomains TLRs and NODs, decrease inflammation and HS-CRP, lower BP, decrease LDL particle number (LDL P), increase LDL and high density lipoprotein (HDL) size, improve glycation parameters, decrease immune vascular dysfunction, decrease CHD plaque formation, improve CHD and CHF symptoms and outcomes
The goal is to use a systematic (dynamic systems biology), functional and metabolic medicine approach to establish cardiovascular ecology, balance, and allostasis (achieve stability through change), and minimize chronic internal and external cardiovascular stressors, mediators, and risk factors that insult the blood vessel. An attempt should be made to reduce the allostatic load, and prevent, regulate, and treat the “abnormal” downstream finite responses.
The polygenetic codes for CVD identify 30 separate loci that are associated with MI and CHD, but only a minority of those 30 loci has anything to do with the top five cardiovascular risk factors. The majority of those loci deal directly with inflammatory pathways. Evaluation and treatment of only the top five risk factors and how they interact with our genome will likely not reduce CVD, and the CHD gap will persist.
Endothelial dysfunction has been hypothesized to be the link between post-prandial state and atherosclerosis and vascular disease. Ingestion of sodium chloride, refined carbohydrates, and foods containing saturated fats and trans fats, lead to prolonged increases in glucose and triglycerides, vascular endotoxemia, inflammation, oxidative stress, and immune dysfunction. Furthermore, these responses may be perpetuated long after the original insult with a heightened continued inflammatory response (metabolic memory). Fortunately, studies have shown that eating a diet rich in low-glycemic foods, monounsaturated and polyunsaturated fats, polyphenols, and antioxidants can help to prevent post-prandial endothelial dysfunction.Early evidence of CVD in the form of fatty streaks has been documented in children in the first and second decades of life (Figure 3). The vascular disease is sub-clinical for 10–30 years or more prior to any cardiovascular event. Endothelial dysfunction is the earliest functional abnormality, followed by changes in arterial compliance, stiffness, and elasticity. It is important to begin using technologies that allow earlier identification of cardiovascular dysfunction before any structural changes have occurred.
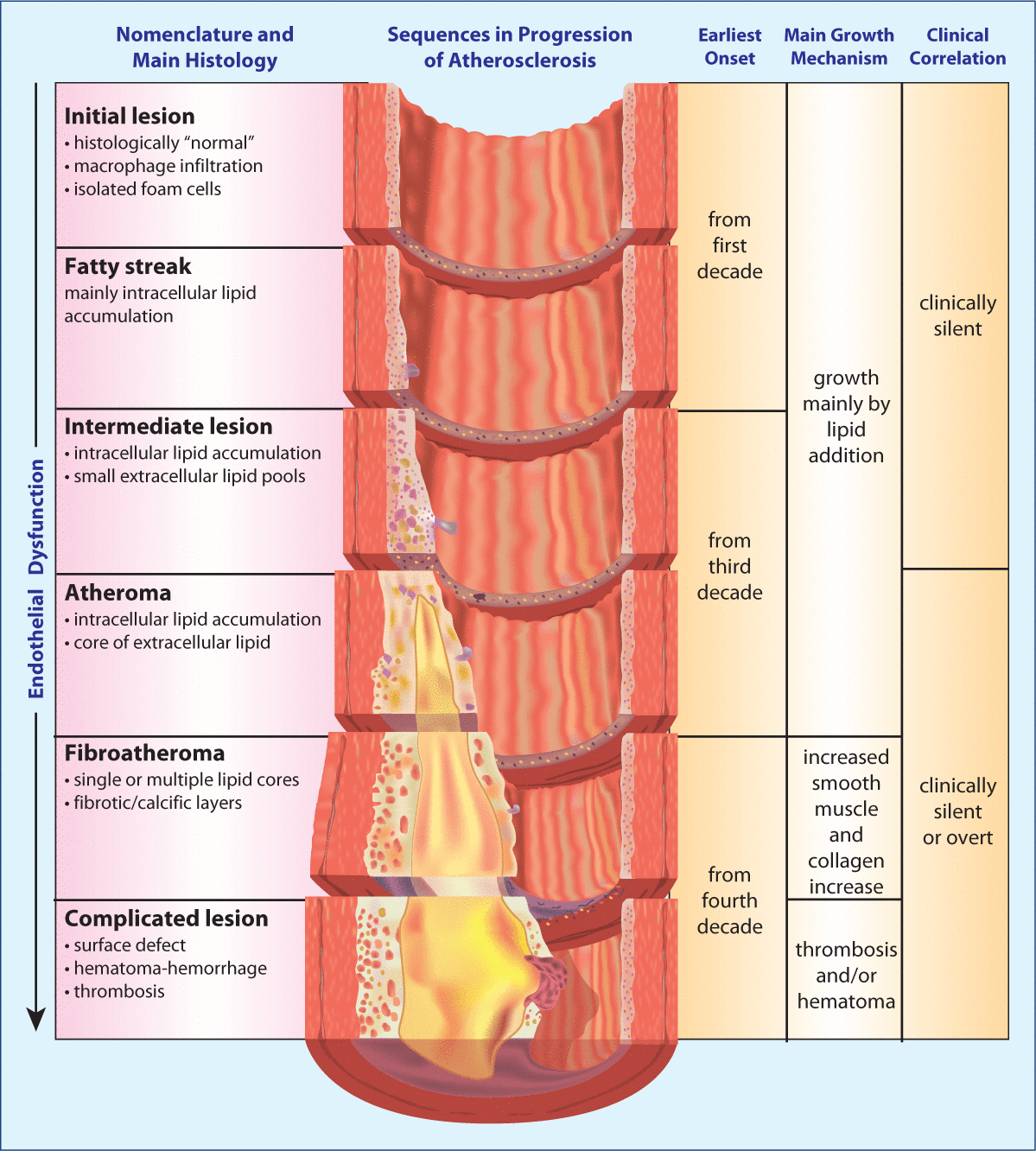
Figure 3: Progression of atherosclerosis.
Figure 4 illustrates the vessel changes that occur as CHD progresses. On the left is a fairly normal artery. In the middle, the CHD has progressed from minimal to moderate with the subendothelium layer becoming thick, but the lumen is still the same size. This extraluminal plaque and inflammation could be observed with electron beam tomography or computed tomography (CT) angiogram but missed by conventional coronary arteriogram (Figure 5). In the image on the right in Figure 4 there is extensive extraluminal and intraluminal disease.
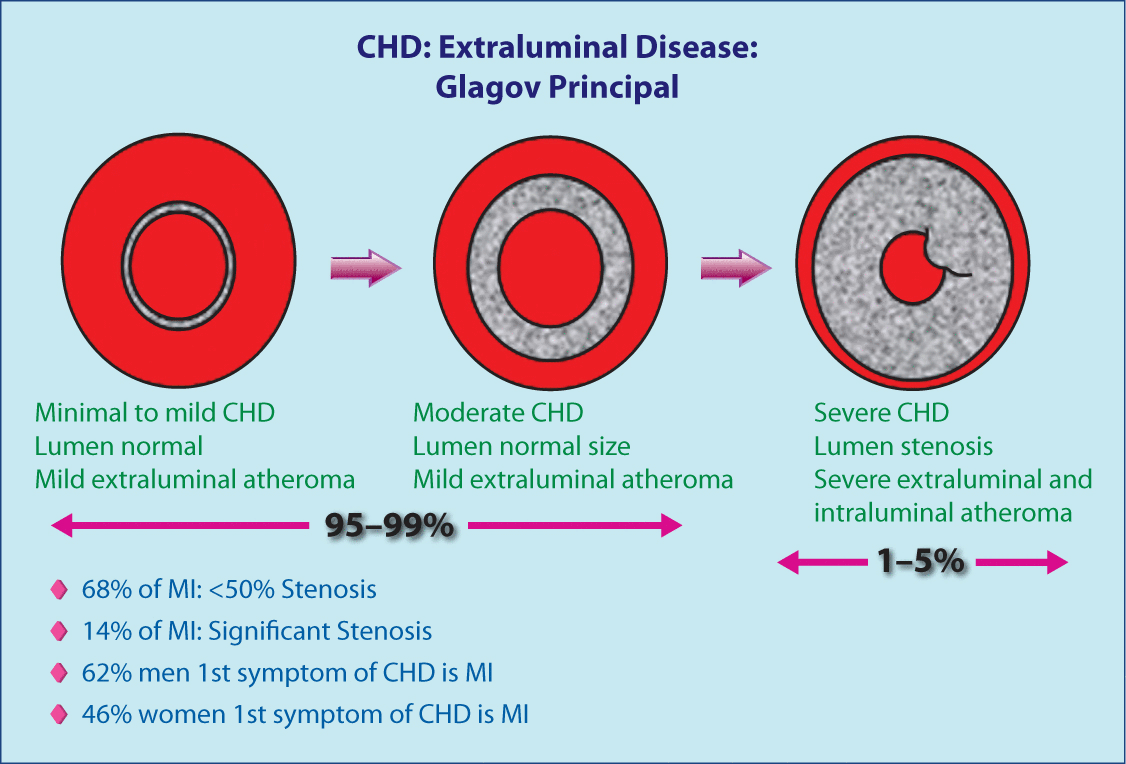
Illustration of the vessel changes that occur as CHD progresses.
CHD, coronary heart disease.
From Nissen SE. Manag Care. 2002;11 (10 Suppl):8–14. Reproduced with permission.
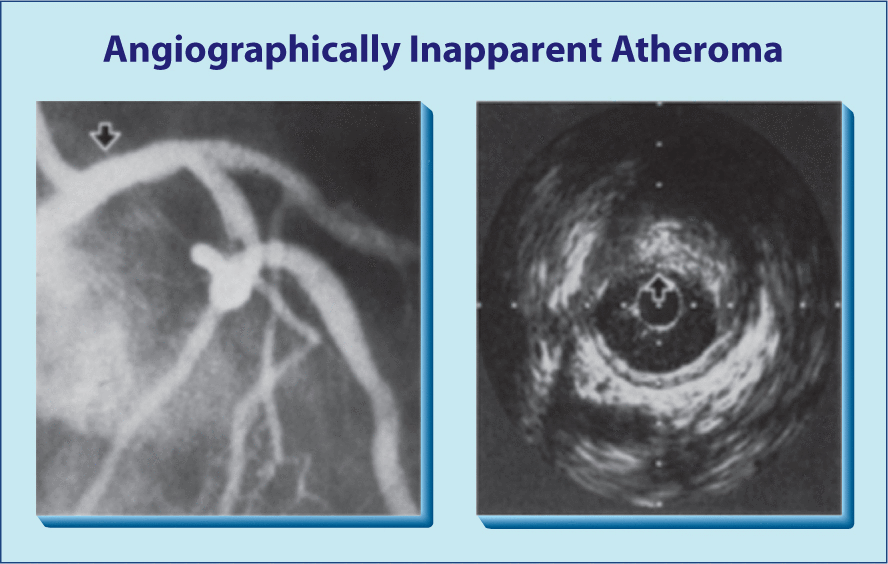
Coronary heart disease that is not detectable by angiogram (left) is clearly evident using computed tomography (right).
From Nissen SE. Manag Care. 2002;11 (10 Suppl):8–14. Reproduced with permission.
Lack of the proper type of imaging, ignoring the majority of the other 400 or so CHD risk factors and not properly evaluating the top five risk factors are some of the reasons for the persistence of the CHD gap. For example, only a 24-hour ambulatory blood pressure monitor can identify specific BP risks for CVD such as nocturnal BP, dipping, non-dipping, BP surges, BP load and BP variability. Non-dipping is defined as a <10% reduction in BP at night. Nocturnal BP is the primary determinate of CVD related to BP measurements. The BP load is the number of BP readings >140/90 mmHg in 24 hours. The normal BP load is <15% of the total BP readings. BP surges that are high and rapid during the early morning between 3 and 9 a.m., as well as labile or variable BP will increase CVD.Excessive dipping is associated with an increased risk of ischemic stroke and reverse dipping is associated with an increased risk of intracerebral hemorrhage. Nocturnal BP is more clinically important than day BP (27/15 mmHg difference is optimal). Furthermore, morning BP surges (level and rapidity) increase the risk of ischemic stroke, MI, and left ventricular hypertrophy.Hypertension is not a disease, it is a marker for vascular dysfunction. Therefore it is crucial that it is correctly identified. The following points should always be considered when evaluating BP:
- Normal BP is 120/80 mmHg, but there is a continuum of risk for CVD starting at 110/70 mmHg;
- Each increase of 20/10 mmHg doubles cardiovascular risk);
- Before age 50, the diastolic BP predicts risk best;
- After age 50, the systolic BP predicts risk best);
- 24-hour ambulatory BP monitoring is more accurate than office BP measurements and should be the standard of care for defining BP and CVD risk;
- Mercury cuffs are best. Electronic arm cuffs are good. Do not use wrist or finger monitors;
- BP load: this should be <15% higher than 140/90 mmHg.
Dyslipidemia is another one of the top five cardiovascular risk factors; however, proper measurement using advanced lipid profiles is not often obtained to verify risk and optimal treatment. An advanced lipid profile, though not routinely used in routine clinical practice, is emerging in research as it measures several risk factors associated with CVD, including:
- LDL-C total
- LDL-P (particle number, drives CHD risk);
- LDL size (dense type B versus large type A);
- modified LDL (oxidized, glycated, glyco-oxidized and acetylated);
- antibodies to oxidized LDL and modified LDL;
- if apolipoprotein (APO) B is elevated;
- APO B antibodies and immune complexes;
- Lp(a);
- HDL-C total;
- HDL-P (particle number);
- HDL size (large 2b versus small type 3);
- dysfunctional HDL;
- pro-inflammatory and pro-atherogenic HDL;
- myeloperoxidase (MPO) and dysfunctional APO A;
- low APO A;
- low paraoxonase (PON)-1 and PON-2;
- increased APO-CIII;
- serum free fatty acids;
- very LDL (VLDL) and triglyceride (TG) total;
- large VLDL;
- VLDL-P particle number;
- remnant particles.
The primary factor driving cardiovascular risk related to LDL cholesterol is the number of LDL particles (LDL-P and APO B particles). HDL-P (particle number) is most protective, with larger HDL type 2b providing the second most important protective mechanism. Larger numbers and sizes of HDL molecules are more efficient at reverse cholesterol transport, and are more protective to the vascular system in numerous other ways. It is also important to analyze dysfunctional HDL. Patients who have an HDL level of 85 mg/dl or more often have dysfunctional HDL that is not even protective. VLDL, triglycerides and remnant particles are very atherogenic and thrombogenic.
A fasting blood sugar (FBS) level of >75 mg/dl increases CHD by 1% per increase of 1 mg/dl, and induces endothelial dysfunction. If a patient has an FBS of 100 mg/dl (often considered a normal level) the risk of CHD is increased by 25%. A 2-hour glucose tolerance test (GTT) measuring >110 mg/dl increases CHD by 2% per 1 mg/dl increase in glucose. The current definition of an abnormal 2-hour GTT is >140 mg/dl. If a patient’s result is 140 mg/dl, which again is currently classed as “normal,” CHD and MI are increased by 60%. Hyperinsulinemia is also an independent risk factor for CHD. Insulin resistance creates inflammation, reduces nitric oxide levels, and causes endothelial dysfunction and vascular disease through the mitogen activated protein kinase (MAPK) pathway, which is atherogenic and induces hypertension as opposed to the phosphatidylinositol 3-kinase (PI3K) pathway, which is anti-inflammatory, anti-hypertensive and anti-atherogenic. It is important to measure all glycation parameters, including fasting glucose, 2-hour GTT, insulin levels, C-peptide and pro-insulin, depending on the clinical setting.
Obesity with increased levels of inflammatory and oxidative stress-related adipokines contribute to CHD. Measurement of not just body weight, but body mass index and body composition with visceral fat and lean body mass will help predict CHD risk.


Fortunately, there are a number of non-invasive tests to determine vascular pathology before it actually starts. Emerging technologies, particularly EndoPAT, a post-brachial artery study, which is very accurate at assessing endothelial function and diagnosing endothelial dysfunction, computerized arterial pulse wave analysis (CAPWA) for endothelial function and arterial compliance, carotid intimal medial thickness (IMT), magnetic cardiography (MCG), and cardiac CT angiograms for calcium score deserve consideration (Table 1). The EndoPAT is a cost effective and noninvasive test that identifies early endothelial dysfunction to predict future CVD and CHD. Further research is needed prior to wide implementation. This test, in addition to 24-hour BP, advanced lipid testing, and glycation measurements, provide cost-effective ways to evaluate several key biomarkers associated with risk in the cardiovascular patient. Numerous other CHD risk factors are listed below in Table 2. Some of the most neglected and important CHD risk factors to evaluate include gender-specific hormones, thyroid function, toxins, homocysteine and vitamin D.


Non-invasive vascular testing for cardiovascular disease.
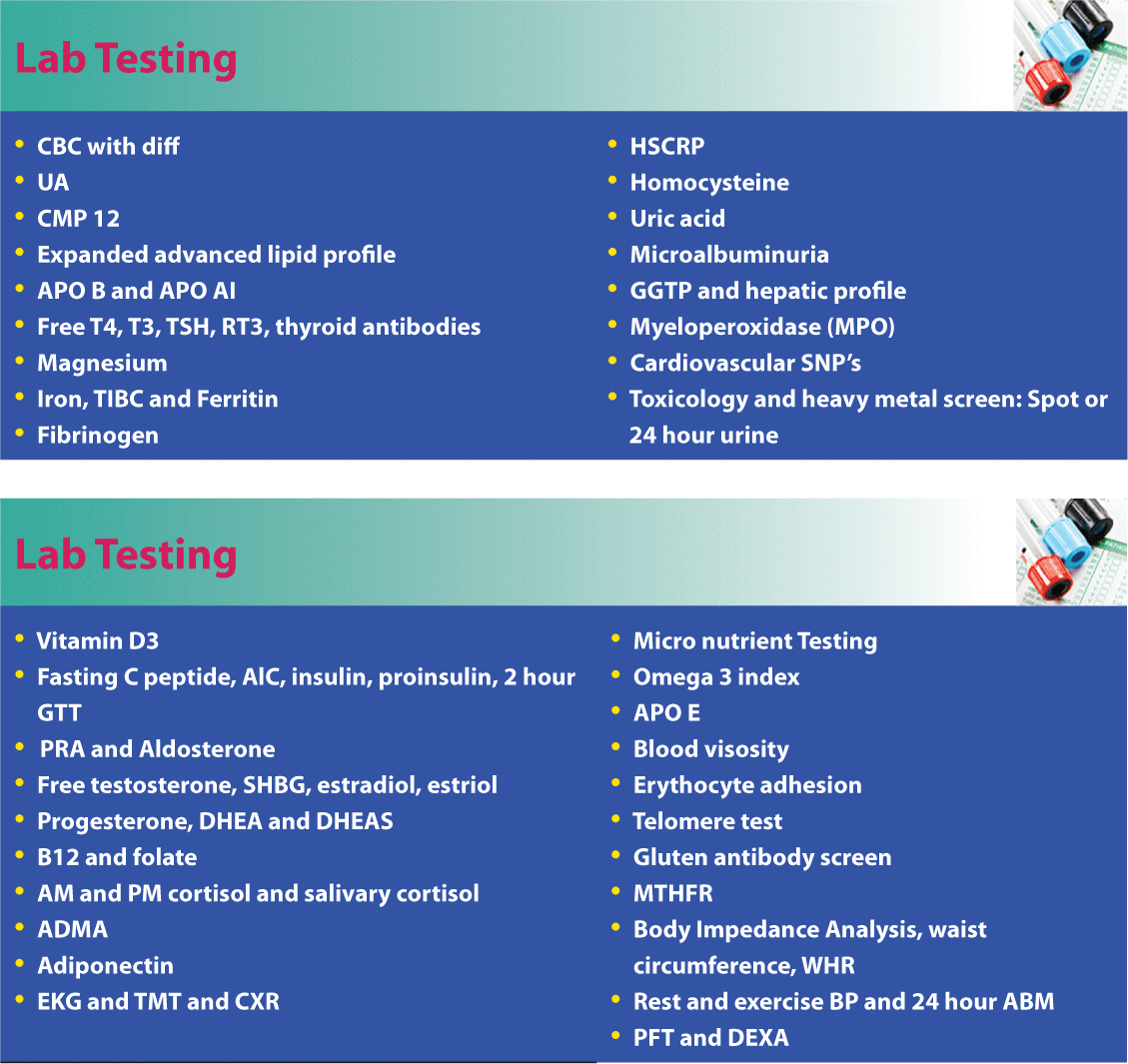
Other comprehensive lab tests for cardiovascular disease.
CONCLUSIONS
The top five cardiovascular risk factors, as they are currently defined, are not an adequate explanation for CHD. In order to close the CHD gap, the top five risk factors must be better defined while assessing the other 395 risk factors and mediators. Early detection and aggressive prevention and treatment of vascular disease are needed before any structural changes occur. To do this we need to utilize new laboratory techniques, such as the advanced lipid profiles, 24-hour BP monitoring, and specific tests to identify inflammation (such as HS-CRP), oxidative stress (such as oxidized LDL and myeloperoxidase), and immune vascular dysfunction. In addition, vascular translational medicine will need to be evaluated with new imaging technologies, such as EndoPAT, CAPWA, carotid IMT, MCG and CT angiogram.
In order to truly revolutionize the treatment of CVD, new therapies will need to involve management of the pathophysiologic risk factors, mediators and their downstream effects, as well as the finite vascular responses. This will be achievable by using a combination of targeted personalized treatments with genomics, proteomics, metabolomics, nutrition, nutraceutical supplements, vitamins, minerals, anti-oxidants, anti-inflammatory agents, anti-immunological agents, and pharmacologic agents. Future studies must begin to measure all of the pertinent risk factors that have been reviewed here to correlate their direct relationship with CHD. Only by addressing all of these factors will we be able to decrease or halt subsequent vascular damage.
DISCLOSURE OF INTERESTS
Dr. Houston has nothing to disclose.
REFERENCES
- Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. INTERHEART Study Investigators.
- World Health Organization. Cardiovascular diseases (CVDs). Fact Sheet No. 317. Accessed August 07, 2013. 2013. Available at:http://www.who.int/mediacentre/factsheets/fs317/en/ .
- Houston MC. What Your Doctor May Not Tell You About Heart Disease. The Revolutionary Book that Reveals the Truth Behind Coronary Illnesses and How You Can Fight Them. Grand Central Life and Style. New York: Hachette Book Group, 2012.
- O’Donnell CJ, Nabel EG. Genomics of cardiovascular disease.
- Houston MC. Nutrition and nutraceutical supplements in the treatment of hypertension.
- ACCORD Study Group, Gerstein HC, Miller ME, et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes.
- Harper CR, Jacobson TA. Treating isolated low high-density lipoprotein cholesterol: prescient or premature?
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III).
- Youssef-Elabd EM, McGee KC, Tripathi G, et al. Acute and chronic saturated fatty acid treatment as a key instigator of the TLR-mediated inflammatory response in human adipose tissue, in vitro.
- El Khatib N, Génieys S, Kazmierczak B, Volpert V. Mathematical modelling of atherosclerosis as an inflammatory disease.
- Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. The Sixth Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC VI).
- National Cholesterol Education Program (NCEP) Expert Panel on Detection Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report.
- American Diabetes Association. Standards of medical care in diabetes.
- Dzau VJ, Antman EM, Black HR, et al. The cardiovascular disease continuum validated: clinical evidence of improved patient outcomes: part I: pathophysiology and clinical trial evidence (risk factors through stable coronary artery disease).
- Dzau VJ, Antman EM, Black HR, et al. The cardiovascular disease continuum validated: clinical evidence of improved patient outcomes: part II: clinical trial evidence (acute coronary syndromes through renal disease) and future directions.
- Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, Clark LT, Hunninghake DB, Pasternak RC, Smith SC Jr, Stone NJ. Coordinating Committee of the National Cholesterol Education Program. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines.
- Port S, Garfinkel A, Boyle N. There is a non-linear relationship between mortality and blood pressure.
- Brunner EJ, Shipley MJ, Witte DR, Fuller JH, Marmot MG. Relation between blood glucose and coronary mortality over 33 years in the Whitehall Study.
- Kannel WB, McGee D, Gordon T. A general cardiovascular risk profile: the Framingham Study.
- Houston MC, Bestermann WH, Ferrario CM, et al. Addressing the global cardiovascular risk of hypertension, dyslipidemia, diabetes mellitus, and the metabolic syndrome in the southeastern United States: Part I.
- Houston MC.
- Della Rocca DG, Pepine CJ. Endothelium as a predictor of adverse outcomes.
- Houston M. The role of nutraceutical supplements in the treatment of dyslipidemia.
- Osterud B, Bjorklid E. Role of monocytes in atherogenesis.
- Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease.
- Esper RJ, Nordaby RA, Vilariño JO, Paragano A, Cacharrón JL, Machado RA. Endothelial dysfunction: a comprehensive appraisal.
- Libby P. Inflammation and cardiovascular disease mechanisms.
- Brown NJ. Aldosterone and vascular inflammation.
- Pauletto P, Rattazzi M. Inflammation and hypertension: the search for a link.
- Tian N, Penman AD, Mawson AR, Manning RD Jr, Flessner MF. Association between circulating specific leukocyte types and blood pressure: the atherosclerosis risk in communities (ARIC) study.
- Muller DN, Kvakan H, Luft FC. Immune-related effects in hypertension and target-organ damage.
- Herrada AA, Campino C, Amador CA, Michea LF, Fardella CE, Kalergis AM. Aldosterone as a modulator of immunity: implications in the organ damage.
- Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension.
- Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension.
- Schiffrin EL. T lymphocytes: a role in hypertension?
- Lundberg AM, Yan ZQ. Innate immune recognition receptors and damage-associated molecular patterns in plaque inflammation.
- Speer T, Rohrer L, Blyszczu P, Shroff R, et al. Abnormal high-density lipoprotein induces endothelial dysfuntion via activation of Tol-like receptor-2.
- Falck-Hansen M, Kassiteridi C, Monaco C. Toll-Like Receptors in Atherosclerosis.
- Opitz B, Eitel J, Meixenberger K, Suttorp N. Role of Toll-like receptors, NOD-like receptors and RIG-I-like receptors in endothelial cells and systemic infections.
- Zhao L, Lee JY, Hwang DH. Inhibition of pattern recognition receptor-mediated inflammation by bioactive phytochemicals.
- Layne J, Majkova Z, Smart EJ, Toborek M, Hennig B. Caveolae: a regulatory platform for nutritional modulation of inflammatory diseases.
- Ansar S, Koska J, Reaven PD. Postprandial hyperlipidemia, endothelial dysfunction and cardiovascular risk: focus on incretins.
- Mah E, Bruno RS. Postprandial hyperglycemia on vascular endothelial function: Mechanisms and consequences.
- Folsom AR, Parker ED, Harnack LJ. Degree of concordance with DASH diet guidelines and incidence of hypertension and fatal cardiovascular disease.
- Stampfer MJ, Hu FB, Manson JE, Rimm EB, Willett WC. Primary prevention of coronary heart disease in women through diet and lifestyle.
- Fan J, Song Y, Wang Y, Hui R, Zhang W. Dietary glycemic index, glycemic load, and risk of coronary heart disease, stroke, and stroke mortality: a systematic review with meta-analysis.
- Solomon TP, Haus JM, Kelly KR, Cook MD, Riccardi M, Rocco M, Kashyap SR, Barkoukis H, Kirwan JP. Randomized trial on the effects of a 7-d low-glycemic diet and exercise intervention on insulin resistance in older obese humans.
- Qi L, Hu FB. Dietary glycemic load, whole grains, and systemic inflammation in diabetes: the epidemiological evidence.
- Kario K, Shimada K, Pickering TG. Abnormal nocturnal blood pressure falls in elderly hypertension: clinical significance and determinants.
- Hermida RC, Ayala DE, Mojón A, Fernández JR. Blunted sleep-time relative blood pressure decline increases cardi-ovascular risk independent of blood pressure level—the “normotensive non-dipper” paradox.
- International Society for Chronobiology; American Association of Medical Chronobiology and Chronotherapeutics; Spanish Society of Applied Chronobiology, Chronotherapy, and Vascular Risk; Spanish Society of Atherosclerosis; Ro-manian Society of Internal Medicine, Hermida RC, Smolensky MH, Ayala DE, Portaluppi F. 2013 ambulatory blood pressure monitoring recommendations for the diagnosis of adult hypertension, assessment of cardiovascular and other hypertension-associated risk, and attainment of therapeutic goals.
- Franco V, Oparil S, Carretero OA. Hypertensive therapy: Part I.
- Franklin SS, Larson MG, Khan SA, Wong ND, Leip EP, Kannel WB, Levy D. Does the relation of blood pressure to coronary heart disease risk change with aging? The Framingham Heart Study.
- O’Brien E, Waeber B, Parati G, Staessen J, Myers MG. Blood pressure measuring devices: recommendations of the European Society of Hypertension.
- Redon J. The importance of 24-hour ambulatory blood pressure monitoring in patients at risk of cardiovascular events.
- Fazio S, Linton MF. High-density lipoprotein therapeutics and cardiovascular prevention.
- van der Steeg WA, Holme I, Boekholdt SM, et al. High-density lipoprotein cholesterol, high-density lipoprotein particle size, and apolipoprotein A-I: Significance for cardiovascular risk: The IDEAL and EPIC-Norfolk studies.
- Cromwell WC, Barringer TA. Low-density lipoprotein and apolipoprotein B: clinical use in patients with coronary heart disease.
- Mackey RH, Greenland P, Goff DC Jr, Lloyd-Jones D, Sibley CT, Mora S. High-density lipoprotein cholesterol and particle concentrations, carotid atherosclerosis, and coronary events: MESA (multi-ethnic study of atherosclerosis).
- Houston M.
- Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Kuvin JT, Lerman AJ. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia.
- Rozanski A, Gransar H, Shaw LJ, et al. Impact of coronary artery calcium scanning on coronary risk factors and downstream testing the EISNER (Early Identification of Subclinical Atherosclerosis by Noninvasive Imaging Research) prospective randomized trial.
- Kandori A, Ogata K, Miyashita T, et al. Subtraction magnetocardiogram for detecting coronary heart disease.
- Moerland M, Kales AJ, Schrier L, van Dongen MG, Bradnock D, Burggraaf J. Evaluation of the EndoPAT as a tool to assess endothelial function. Int J Vasc Med . 2012; 2012:904141.