Abstract
There have been recent advances in understanding of the local control of thyroid activity and metabolism, including deiodinase activity and thyroid hormone membrane transport. The goal of this review is to increase the understanding of the clinical relevance of cellular deiodinase activity. The physiologic significance of types 1, 2 and 3 deiodinase (D1, D2 and D3, respectively) on the intracellular production of T3 are discussed along with the importance and significance of the production of reverse T3. The difference in the pituitary and peripheral activity of these deidoidinases under a wide range of common physiologic conditions results in different intracellular T3 levels in the pituitary and peripheral tissues, resulting in the inability to detect low tissue levels of thyroid hormone in peripheral tissues with TSH testing. This review demonstrates that extreme caution should be used in relying on TSH or serum thyroid levels to rule out hypothyroidism in the presence of a wide range of conditions, including physiologic and emotional stress, depression, dieting, obesity, leptin insulin resistance, diabetes, chronic fatigue syndrome, fibromyalgia, inflammation, autoimmune disease, or systemic illness, as TSH levels will often be normal despite the presence of significant hypothyroidism. The review discusses the significant clinical benefits of thyroid replacement in such conditions despite having normal TSH levels and the superiority of T3 replacement instead of standard T4 therapy.
INTRODUCTION
To accurately assess thyroid function, it must be understood that deiodinase enzymes are essential control points of cellular thyroid activity that determine intracellular activation and deactivation of thyroid hormones. This local control of cellular thyroid levels is mediated through three different deiodinase enzymes present in different tissues in the body; type I deiodinase (D1) and type II deiodinase (D2) increase cellular thyroid activity by converting inactive thyroxine (T4) to the active triiodothyronine (T3) while type III deiodinase (D3) reduces cellular thyroid activity by converting T4 to the anti-thyroid reverse T3 (reverse T3)(see Figure 1).
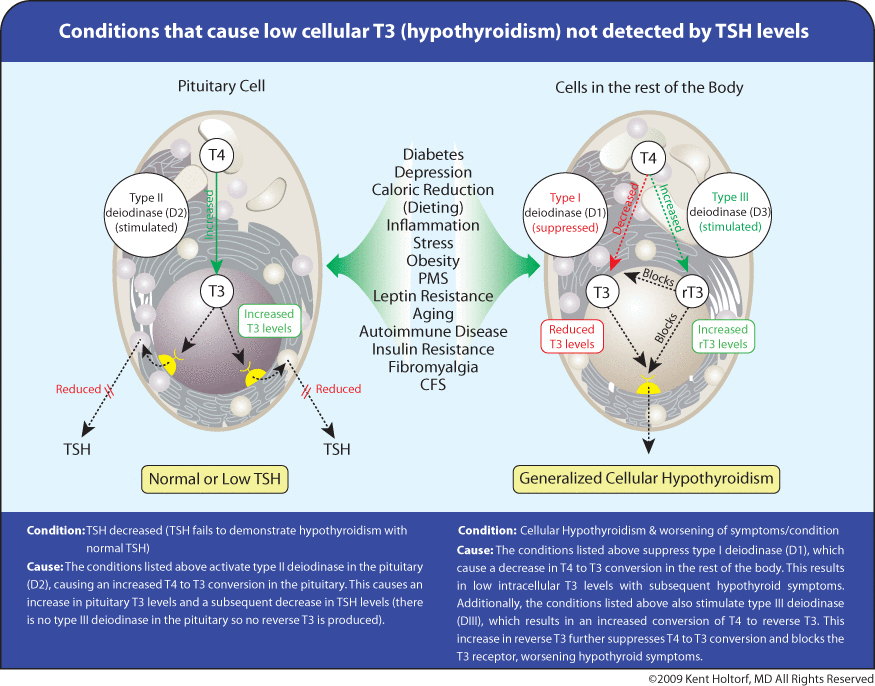
The activity of each type of deiodinase enzyme changes in response to differing physiologic conditions, and this local control of intracellular T4 and T3 levels results in different tissue levels of T4 and T3 under different conditions. Because it is the activity of these deiodinases and transport of T4 and T3 into the cell that determines tissue and cellular thyroid levels and not serum thyroid levels, serum thyroid hormone levels may not necessarily predict tissue thyroid levels under a variety of physiologic conditions.
DEIODINASE TYPE I (D1)
D1 converts inactive T4 to active T3 throughout the body, but D1 is not a significant determinant of pituitary T4 to T3 conversion, which is controlled by D2. D1 but not D2 is suppressed and down-regulated (decreasing T4 to T3 conversion) in response to physiologic and emotional stress;depression; dieting; weight gain and leptin resistance; insulin resistance, obesity and diabetes; inflammation from autoimmune disease or systemic illness; chronic fatigue syndrome and fibromyalgia; chronic pain; and exposure to toxins and plastics. In the presences of such conditions there are reduced tissue levels of active thyroid in all tissues except the pituitary. The reduced thyroid tissue levels with these conditions is often quoted as a beneficial response that lowers metabolism and thus does not require treatment, but there is no evidence to support such a stance, while there is significant evidence demonstrating it is a detrimental response.
In addition, D1 activity is also lower in females, making women more prone to tissue hypothyroidism, with resultant depression, fatigue, fibromyalgia, chronic fatigue syndrome, and obesity despite having normal TSH levels.
DEIODINASE TYPE II (D2)
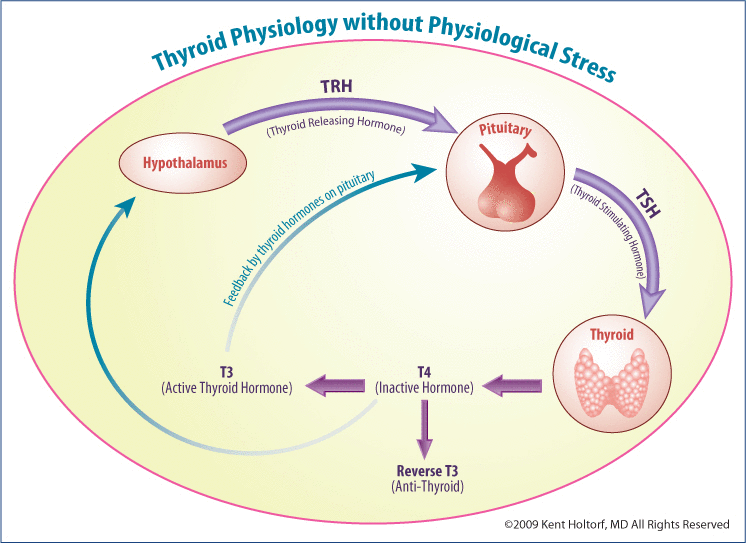
Thyroid stimulating hormone (TSH) is produced in the pituitary and is regulated by intra-pituitary T3 levels, which often do not correlate or provide an accurate indicator of T3 levels in the rest of the body. Using the TSH as a indicator for the body’s overall thyroid status assumes that the T3 levels in the pituitary directly correlate with that of other tissues in the body and that changes directly correlate with that of T3 in other tissue of the body under a wide range of physiologic conditions. This, however, is shown not to be the case; the pituitary is different than every other tissue in the body.
Due to a unique make-up of deiodinases in the pituitary, it will respond differently and often opposite to that of every other tissue in the body. Numerous conditions result in an increase in pituitary T3 levels while simultaneously suppressing cellular T3 levels in the rest of the body, making the pituitary, and thus the TSH, a poor indicator for tissue thyroid levels in the rest of the body under numerous physiologic conditions.
In addition to having a unique make-up of deiodinases, the pituitary also contains unique membrane thyroid transporters and thyroid receptors. As opposed to the rest of the body that is regulated by both D1 and D3, the pituitary contains little D1 and no D3; so pituitary T3 levels are determined by D2 activity, which is 1000 times more efficient at converting T4 to T3 than the D1 enzyme present in the rest of the body and is much less sensitive to suppression by toxins and medications. Though D2 activity is present in human skeletal muscle (unexpected from studies in rats), there is less D1 and D3 present in the pituitary than in the other tissues of the body. In the pituitary, 80–90% of T4 is converted to T3, while only about 30–50% of T4 in the peripheral tissue is converted to active T3. This is due to the inefficiency of D1 and the presence of D3 in all tissues of the body except the pituitary that competes with D1 and converts T4 to reverse T3.
Additionally, D2 also has an opposite response from that of D1 to physiologic and emotional stress, depression, both dieting and weight gain, PMS, diabetes, leptin resistance, chronic fatigue syndrome, fibromyalgia, inflammation, autoimmune disease, and systemic illness. D2 is stimulated and up-regulated (increased activity) in response to such conditions, increasing intra-pituitary T4 to T3 conversion while the rest of body suffers from diminished levels of active T3. This causes the TSH to remain normal despite the fact that there is significant cellular hypothyroidism present in the rest of the body.
Thus, the pituitary levels are under completely different physiologic control and T3 levels will always be significantly higher than anywhere else in the body. Consequently, if the TSH is elevated, even mildly, it is clear that many tissues of the body will be deficient in T3; but due to the different physiology, a normal TSH cannot be used as a reliable indicator for normal T3 levels in the rest of the body.
Different thyroid levels and conditions will have different effects on the T3 levels in the pituitary than in the rest of the body, resulting in different T3 levels in the pituitary and the rest of the body, making the TSH unreliable under numerous circumstances. For instance, as the levels of T4 decline, as in hypothyroidism, the activity of the D2 increases and is able to partially compensate for the reduction in serum T4. On the other hand, with reduced T4 levels, the activity and efficiency of D1 decreases resulting in a reduction in cellular T3 levels while the TSH remains unchanged due to the ability of the pituitary D2 to compensate for the diminished T4.
As stated above, this lack of correlation of TSH and peripheral tissue levels of T3 is dramatically worsened in numerous conditions. These include chronic emotional or physical stress, chronic illness, diabetes, insulin resistance, obesity, leptin resistance, depression, chronic fatigue syndrome, fibromyalgia, PMS, and both dieting and weight gain. In such conditions, tissue levels of T3 are shown to drop dramatically out of proportion with serum T3 levels. While serum T3 levels may drop by 30%, which is significant but still may be in the so-called “normal range,” tissue T3 may drop by 70–80%, resulting in profound cellular hypothyroidism with normal serum TSH, T4, and T3 levels. Consequently, in the presence of such conditions, the TSH is a poor indicator for peripheral thyroid levels and a normal TSH should not be considered a reliable indicator for an individual being euthyroid (normal thyroid), especially in the presence of symptoms consistent with thyroid deficiency.
Doctors in the thyroid division of the Department of Medicine at Brigham and Women’s Hospital and Harvard Medical School investigated how the pituitary’s unique deiodinase makeup responds differently than the tissues of the rest of body and how the pituitary is a poor indicator for thyroid levels in the rest of the body. In their review published in Endocrine Reviews, the authors state, “The approximately 1000-fold lower Km of D2 than D1 [D2 is 1000 times more efficient] may give this enzyme a major advantage in terms of extrathyroidal T3 production... The free T3 concentration in different tissues varies according to the amounts of hormone transported and the activity of the tissue deiodenases. As a result, the impact of the plasma thyroid hormones on target tissues is not the same in every tissue.”
In the journal Endocrinology, Lim et al. measured peripheral (liver) and pituitary levels of T3 in response to induced chronic illness. They found that pituitary T3 and TSH levels remained unchanged while the peripheral tissues were significantly reduced. The authors summarize their findings by stating:
“The reduction in hepatic nuclear T3 content and T3-Cmax in the Nx2 rats is consistent with the presence of selective tissue deficiency of thyroid hormones. The pituitary, however, had normal T3 content, suggesting a dissociation in thyroid hormone-dependent metabolic status between peripheral tissue (liver) and the pituitary. This explains the failure to observe an increase in serum TSH level, a manifestation of reduced intracellular rather than serum T3 concentration…Most interesting, we found that, in contrast to the liver, the pituitary of the Nx rats was not deprived of thyroid hormone. This finding offers a convincing explanation of the failure to observe an increase of serum TSH when illness or stress-induced reduction of hepatic T4 5′-monodeiodination causes a fall in serum t3 concentration.”
In the New England Journal of Medicine, Larsen et al. summarize the fact that the pituitary has a unique composition of deiodinases that is not present in any other tissue in the body, making the pituitary T3 levels, and thus the TSH, a poor indicator for tissue T3 in the rest of the body – stating that the TSH cannot be reliably used as a marker of thyroid status in the rest of the body.
“Changes in pituitary conversion of T4 to T3 are often opposite of those that occur in the liver and kidney under similar circumstances. The presence of this pathway of T3 production indicates that the pituitary can respond independently to changes in plasma levels of T4 and T3…Given these results, it is not surprising that a complete definition of thyroid status requires more than the measurement of the serum concentrations of thyroid hormones. For some tissues, the intracellular T3 concentration may only partially reflect those in the serum. Recognition that the intracellular T3 concentration in each tissue may be subject to local regulation and an understanding of the importance of this process to the regulation of TSH production should permit a better appreciation of the limitations of the measurements of serum thyroid hormone and TSH levels.”
DEIODINASE TYPE III (D3)
The pituitary is the only tissue that does not contain D3, which converts T4 to reverse T3 and competes with D1 that converts T4 to T3. Reverse T3 is a competitive inhibitor of T3, blocking T3 from binding to its receptor and blocking T3 effect,reduces metabolism, suppresses D1 and T4 to T3 conversion, and blocks T4 and T3 uptake into the cell, all reducing intracellular T3 levels and thyroid activity. Because many tissues may have abundant D3 levels while the pituitary is uniquely void of D3, the inhibitory effects on the peripheral tissues causing hypothyroidism are not reflected by TSH testing (see Figures 2 and 3).
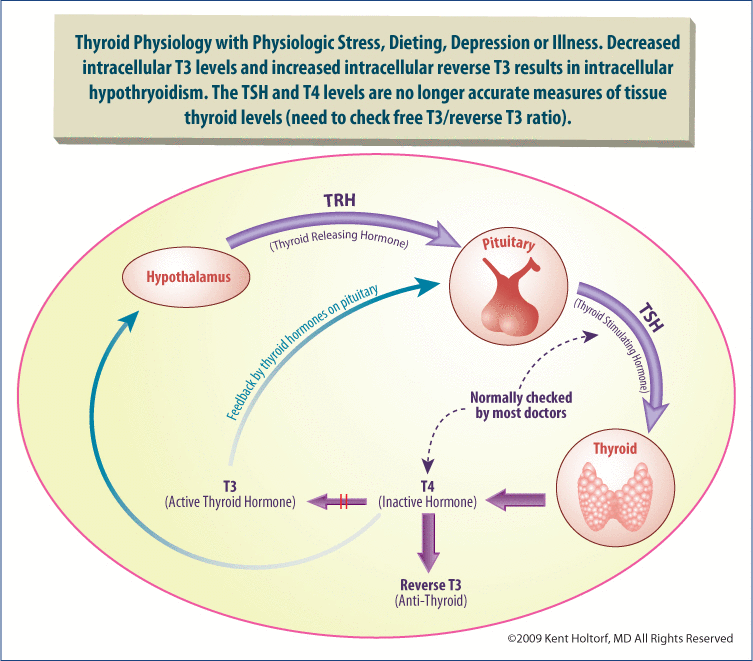
Reverse T3 is present in varying concentrations in different tissues and with different individuals. It is up-regulated with chronic physiologic stress and illness and is an indicator for reduced T4 to T3 conversion and low intracellular T3 levels even if the TSH is normal.
Because increased serum and tissue level of reverse T3 will result in a blocking of the thyroid receptors, even small increases in reverse T3 can result in a significant decrease in thyroid action and result in severe hypothyroidism not detected by standard blood tests. Because any T4 given will contribute to more reverse T3, T4-only preparations should not be considered optimal thyroid replacement in the presence of high or high-normal reverse T3 levels while T3 can be significantly beneficial.
STRESS
Chronic physiologic stress results in decreased D1 activity and an increase in D3 activity, decreasing thyroid activity by converting T4 into reverse T3 instead of T3. Conversely, D2 is stimulated, which results in increased T4 to T3 conversion in the pituitary and reduced production of TSH. The increased cortisol levels seen with stress also contribute to physiologic disconnect between the TSH and peripheral tissue T3 levels. This stress induced reduced tissue T3 level and increased reverse T3 results in tissue hypothyroidism and potential weight gain, fatigue, and depression. This vicious cycle of weight gain, fatigue, and depression that is associated with stress can be prevented with supplementation with timed-released T3 but not T4.
The reduced immunity from chronic stress has been thought to be due to excess cortisol production, but the associated reduction in tissue thyroid levels are shown to play a larger role in the decreased immunity seen with stress, and thyroid supplementation is shown to reverse the stress induced reduction in immunity.
As with stress, treatment with prednisone or other glucocorticoid will suppress D1 and stimulate D3, reducing T4 to T3 conversion and increasing T4 to reverse T3, causing a relative tissue hypothyroidism that is not detected by TSH testing. This low cellular thyroid level certainly contributes to the weight gain and other associated side-effects with such treatment. Thus, in stressed patients or those treated with corticosteroids, there are reduced tissue T3 levels that are not reflected by the TSH level, making the TSH an inappropriate marker for tissue levels of T3.
DEPRESSION
Many depressed and bipolar patients have undiagnosed thyroid dysfunction as the underlying cause or major contributor to their depression. The dysfunction present with these conditions includes down regulation of D1 (reduced T4 to T3 conversion) and reduced uptake of T4 into the cell, resulting in increased serum T4 levels with low intracellular T3 levels and upregulated D3, resulting in elevated reverse T3,which blocks the thyroid effect and is an indicator of reduced transport of T4 into the cell. Additionally, studies show that depressed patients have reduced T4 transport across the blood brain barrier due to a defective transport protein, transthyretin, resulting in significantly reduced thyroid levels in the brains of depressed patients despite “normal” serum levels and standard thyroid tests as well as a reduced TSH response to TRH.
It is not surprising that T4 and T4/T3 combinations may have some benefit in depression; but due to the suppressed T4 to T3 conversion from suppressed D1 and reduced uptake of T4 into the cell and brain, timed-released T3 is significantly more beneficial than T4 or T4/T3 combination supplementation.
In the International Journal of Neruopsychopharmacology, Posternak et al. published a double blind placebo control trial of 50 patients with normal thyroid function as defined by a normal TSH (1.5±0.8). The patients were randomized to receive 25 µg of T3 or placebo in addition to antidepressant therapy. The study found almost a two-fold increase in response rate with T3 and a 4.5 times greater likelihood of experiencing a positive response at any point over a six-week period with the addition of T3. Side effects were higher in placebo group on 10/11 criteria including a significant increase in nervousness with the placebo group.
Kelly et al. investigated the effectiveness of T3 for the treatment of biopolar disorder in patients who had failed to adequately respond to an average of 14 medications used to treat their bipolar disorder. The average dose of T3 used was 90.4 µg (range 13–188 µg). The medication was found to be well tolerated and 84% experienced significant improvement and 33% had a full remission. Again, this is in patients who had not previously responded to numerous medications. One patient who was switched to T4 for cost reasons experienced a return of symptoms, which resolved with the reintroduction of T3. The authors concluded, “Augmentation with supraphysiologic doses of T3 should be considered in cases of treatment resistant bipolar depression….” The authors thanked several doctors who encouraged them to go beyond the traditional 50 µg of T3 because it has helped so many of their patients.
With over 4000 patients, The Star*D Report is the largest trial comparing antidepressant effectiveness for depression. It found that 66% of patients fail to respond to antidepressants or have side-effects severe enough to discontinue use. Of those who do respond, over half will relapse within one year. The trial found that T3 was effective even when other medications – such as citalopram (Celexa), bupropion (Wellbutrin), sertraline (Zolft), venlafaxine (Effexor), or cognitive therapy – were not. It was shown to be 50% more effective, even with the less than optimal dose of 50 µg, under direct comparison with significantly less side effects than commonly used therapeutic approaches with standard antidepressants. The authors included a case study to exemplify the effectiveness of T3, especially when other medications are not:
“Ms. ‘B,’ a 44-year-old divorced white woman, became depressed after losing her job as a secretary in a law firm. She initially sought treatment from her primary care physician and then entered the STAR*D study. Ms. B met criteria for major depressive disorder and generalized anxiety disorder. Her baseline QIDS-SR score was 16. After 12 weeks on citalopram, her QIDS-SR score was 10 [minimal response]. She was then randomly assigned to augmentation with buspirone; she soon experienced gastrointestinal distress, and she stopped taking buspirone after 6 weeks. She elected to try one more augmentation agent and was randomly assigned to T3 augmentation. When she started T3 augmentation, her QIDS-SR score was 12. After 4 weeks, she felt that her mood and energy had lifted substantially. She felt better able to make decisions, organize, and prioritize and felt that she was able and ready to look for another job. ‘I felt as if my brain suddenly had oxygen,’ she said, ‘and everything became clearer.’ After 12 weeks, Ms. B felt back to normal, and her QIDS-SR score was 0 [complete resolution of symptoms].”
With an understanding of thyroid physiology and associated dysfunction that is present in depressed patients, it is clear that timed-released T3 supplementation should be considered in all depressed and bipolar patients despite “normal” serum thyroid levels. Additionally, straight T4 should be considered inappropriate and suboptimal therapy for replacement in such patients.
PAIN
Chronic pain will significantly suppress D1 and upregulate D2, resulting in a reduction in tissue T3 without a change in TSH. Thus, the significant cellular hypothyroidism is not detected by serum TSH and T4 testing. This cellular hypothyroidism, which again is undiagnosed by standard blood tests, increases the risk of the associated fatigue and depression seen with chronic pain.
Narcotic pain medication can, of course, alleviate pain and thus potentially improve the diminished tissue T3 levels seen with chronic pain; but narcotics also suppress D1 but not D2, so such treatment is ineffective at reversing the suppressed tissue T3 levels. Thus, for those with significant chronic pain or using significant amounts of narcotic pain medicine, it must be understood that such a condition is associated with low tissue thyroid levels not detected by standard blood tests. Tolerance to the inhibitory effect of narcotics on TSH secretion and T4 to T3 conversion does not occur. Expert pain specialists understand this and recommend T3 supplementation to patients with significant pain or on narcotic pain medications.
DIETING
Acute or chronic dieting can result in a significant decrease in intracellular and circulating T3 levels by up to 50%, which significantly reduces basal metabolic rate (number of calories burned per day) by 15–40%. With chronic dieting, the thyroid levels and metabolism often do not return to normal levels; the body stays in starvation mode for years with significantly reduced metabolism despite the resumption of normal food intake, making it very difficult to lose or maintain lost weight.
A study by Araujo et al. published in American Journal of Physiology, Endocrinology and Metabolism found that 25 days of calorie restriction (dieting) significantly reduced D1, resulting in reduced T4 to T3 conversion with a 50% reduction in T3. This dramatic reduction in T3 was associated with an increase in D2, so there was no increase in TSH but rather a decrease from an average of 1.20 ng/mL to 0.7 ng/mL, demonstrating the fact that the TSH is a poor marker for tissue T3 levels, especially in a chronically dieting patient.
Fontana et al. found that T3 levels were significantly decreased by 25% in chronically dieting individuals compared to non-dieting individuals with no difference in TSH and T4 (thus undetected by TSH and T4 testing). This clinically significant reduction in T3 levels, potentially causing inability to lose weight or regaining of lost weight, fatigue, and depression, remained in the normal range despite the significant decline, demonstrating the weakness and unreliability of the common use of population reference ranges that consider 95% of the population as “normal”.
A study by Leibel et al. published in the journal Metabolism found that individuals who had lost weight in the past had a significantly lower metabolism than those of same weight who had not gained or lost significant weight in the past. The metabolism in the weight reduced patients was 25% less than an equal weight person who did not lose or gain significant weight in the past and equal to someone who weighed 60% less than they did. Additionally, the reduction was shown to be present years later.
This 25% percent reduction in metabolism equates to an approximate deficit of 500–600 cal per day. Thus, if the previously overweight persons are to maintain the reduced weight they lost, they must either eat 600 cal per day less compared to a person of the same weight who has not had a weight problem or must jog about 1 ½ hours per day to maintain the lost weight. This equates to approximately a pound per week of weight gain, explaining why weight is so quickly gained without continued very strict dieting. So many people who have difficulty keeping weight off don’t eat excessively but are continually told they are eating too much or they need to exercise more by people who have never had a weight problem. They are made to feel it is a character issue and that nobody believes how little food they actually consume. Unless the physiologic thyroid dysfunction is corrected, any diet and exercise strategy is doomed.
Croxson et al. in Journal of Endocrinology and Metabolism found that individuals with a history of intense dieting had dramatic reductions in T4 to T3 conversion with an intracellular deficiency of T3. The inadequacy and inaccuracy of standard TSH and T4 testing was demonstrated, as such testing failed to detect the dramatic reduction in tissue levels of T3 in all of the patients.
INSULIN RESISTANCE/DIABETES/METABOLIC SYNDROME/OBESITY
As with leptin resistance, it has been shown in numerous studies that insulin resistance, diabetes, or metabolic syndrome have associated significant reduction in T4 to T3 conversion, an intracellular deficiency of T3, and an increased conversion of T4 to reverse T3, further reducing intracellular T3 levels. Additionally, the elevated insulin will increase D2 activity and suppress TSH levels, further decreasing thyroid levels and making it inappropriate to use the TSH as a reliable marker for tissue thyroid levels in the presence of elevated insulin levels as occurs with obesity, insulin resistance, or type II diabetes.
Pittman et al. found that normal individuals had a 77% conversion of T4 to T3, while diabetic individuals had a 45% conversion of T4 to T3 and increased T4 to reverse T3. Improvement in glucose levels only slightly increased T4 to T3 conversion to 46%.
Islam et al. investigated the T4 to T3 conversion in 50 diabetic patients compared to 50 non-diabetic controls. There was no difference in TSH and free T4 levels, but the diabetic individuals had significantly decreased free T3 levels (P=0.0001) that averaged 46% less than controls. The FT3/FT4 ratio was 50% less in diabetic patients versus controls. The TSH failed to elevate despite the fact that serum T3 was approximately half of normal.Saunders et al. also found that diabetics had approximately a 50% reduction in T3 levels and significantly increased reverse T3 levels and decreased T3/reverse T3 ratios.
In the International Journal of Obesity, Krotkiewski et al. published the results of their investigation of the impact of supplemental T3 on cardiovascular risk in obese patients to partially reverse the reduced T4 to T3 conversion seen with obesity. Seventy obese patients with “normal” standard thyroid function tests were treated with 20 µg of straight T3 for six weeks. While the dose was not high enough to completely reverse the reduced T4 to T3 conversion seen with obesity, there was a significant reduction in a number of cardiovascular risk factors, including cholesterol and markers for insulin resistance. There were no side-effects in any of the patients. The authors conclude, “T3 may be considered to ameliorate some of the risk factors associated with abdominal obesity, particularly in some subgroups of obese women with a relative resistance to thyroid hormones possibly dependent on decreased peripheral deiodination of thyroxine (T4).”
Thus, replacement with timed-released T3 preparations to normalize the reduced intracellular T3 levels is appropriate in such patients despite so-called “normal” levels while, on the contrary, T4-only preparations do not address the physiologic abnormalities of such patients and should be considered inappropriate replacement for obese patients or those with insulin resistance, leptin resistance, or diabetes, as they do not address the physiologic abnormalities in this group.
LEPTIN
The hormone leptin has been found to be a major regulator of body weight and metabolism. The body secretes leptin as weight is gained to signal the brain (specifically the hypothalamus) that there are adequate energy (fat) stores. The hypothalamus should then stimulate metabolic processes that result in weight loss, including a reduction in hunger, an increased satiety with eating, an increase in resting metabolism, and an increase in lipolysis (fat breakdown). New research has found that this leptin signaling is dysfunctional in the majority of people who have difficultly losing weight or are unable to lose weight.
The problem is not in the production of leptin; studies show that the majority of overweight individuals who are having difficulty losing weight have a leptin resistance, where the leptin is unable to produce its normal effects to stimulate weight loss. This leptin resistance is sensed as starvation, so multiple mechanisms are activated to increase fat stores, rather than burn excess fat stores. Leptin resistance is shown to suppress D1 and stimulate D2, resulting in reduced cellular T3 but a reduction in serum TSH. A study by Cettour-Rose et al. published in American Journal of Physiology, Endocrinology and Metabolism demonstrated that physiologic reversal of leptin resistance restored deiodinase activity except in the presence of elevated reverse T3. Thus, in the presence of elevated leptin level (above 10) there is a reduction of cellular T3 and a suppression of TSH, making the TSH an unreliable indicator of thyroid status, especially when combined with an elevated reverse T3. Thus, for anyone who has difficulty losing weight, a leptin level above 10 demonstrates that low intracellular thyroid level is contributing to this difficulty, especially if combined with a high normal or elevated reverse T3 (above 150).
EXERCISE
It has been shown that women or men who perform more than moderate exercise, especially when associated with dieting, have reduced T4 to T3 conversion and increase reverse T3, counteracting many of the positive effects of exercise in women including weight loss. Consequently, T3 and reverse T3 levels should be evaluated in individuals who exercise and/or diet to better determine cellular thyroid levels, as TSH and T4 would not necessarily reflect tissue levels in such patients.
IRON DEFICIENCY
Iron deficiency is shown to significantly reduce T4 to T3 conversion, increase reverse T3 levels, and block the thermogenic (metabolism boosting) properties of thyroid hormone. Thus, iron deficiency, as indicated by an iron saturation below 25 or a ferritin below 70, will result in diminished intracellular T3 levels. Additionally, T4 should not be considered adequate thyroid replacement if iron deficiency is present.
INFLAMMATION ASSOCIATED WITH COMMON CONDITIONS
The inflammatory cytokines IL-1, Il-6, C-reactive protein (CRP), and TNF-alpha will significantly decrease D1 activity and reduce tissue T3 levels. Any person with an inflammatory condition – including physical or emotional stress, obesity, diabetes,depression, menopause (surgical or natural), heart disease, autoimmune disease (lupus, Hashimoto’s, multiple sclerosis, arthritis, etc.), injury, chronic infection or cancer– will have a decreased T4 to T3 conversion in the body and a relative tissue hypothyroidism. The inflammatory cytokines will, however, increase the activity of D2 and suppress the TSH despite reduced peripheral T3 levels; again, making a normal TSH an unreliable indicator of normal tissue thyroid levels.
There is a direct inverse correlation between CRP and reduced tissue T3, so individuals with elevated CRP (greater than 3 mg/L) or other inflammatory cytokines will have a significant reduction in cellular T3 levels. The suppression of intracellular T3 levels correlates with the degree of elevation of CRP, despite serum thyroid tests being “normal”. Thus, if any inflammation is present, which is found in numerous clinical and subclinical conditions (as above), the body will have lower cellular T3 levels that are often inadequate for optimal functioning; but the pituitary will have increased levels of T3, resulting in a lowering of the TSH that would potentially be inappropriately interpreted as an indication of “normal” thyroid levels.
Thus, any person with an inflammatory condition will have diminished tissue levels of T3 potentially severe enough to cause symptoms, but these symptoms will not be detected by standard thyroid testing. Additionally, due to the reduced T4 to T3 conversion induced by the inflammation in these conditions, effective treatment must include T3 (combination or, ideally, timed-released T3). Also, due to the inflammatory suppression of TSH, not only is a normal TSH necessarily an indication of euthyroidism (normal thyroid), but also a suppressed TSH is not necessarily an indication of excessive thyroid with treatment. Rather, free T3 and reverse T3 levels along with clinical parameters should be used to determine optimal replacement doses of thyroid.
Additionally, inflammation will stimulate D3, producing more reverse T3, further causing cellular hypothyroidism not detected by TSH testing by suppressing intracellular T4 to T3 conversion and blocking the T3 receptor inside the cell.
ENVIRONMENTAL TOXINS
Numerous toxins, including plastics such as bisphenol-A, pesticides, mercury, and flame retardants such as PBDE, are shown to block tissue thyroid receptors and reduce T4 to T3 conversion with resultant low tissue levels of thyroid that are not detected by standard blood tests. In addition to being 1000 times more efficient at converting T4 to T3, D2 is 100- to 1000-fold less sensitive to suppression by toxins or by mineral or hormonal deficiencies. Thus, the D1 in the body is suppressed by toxins, pesticides, and plastics at levels that are hundreds to thousands times lower than required to suppress the D2 in the pituitary. This is proving to be a major problem for the population in general; levels of plastics and other toxins commonly found in individuals (toxins that are considered “normal” exposure) result in reduced levels of T3 in all tissues with the exception of the pituitary, which is resistant to the effect of toxins. Because the pituitary is relatively unaffected, the reduced tissue thyroid levels are not detected by standard TSH testing.
For instance, bisphenol-A, which is ubiquitous in the environment and large amounts of which can leach into food and liquids from plastic water bottles and the lining of aluminum cans, is shown to significantly block thyroid activity in all tissues except the pituitary, potentially contributing to or causing weight gain, fatigue, and depression but not detected by TSH testing. Levels of a number of thyroid blocking toxins, including bisphenol-A and PBDEs, are significantly higher in individuals in the United States (PBDEs being especially high in California), resulting in reduced T3 effect in all tissues in almost all individuals in the United States compared to the rest of the world that is not detected by standard thyroid testing. This is potentially a significant contributor to the epidemic of obesity and depression in the US.
TESTOSTERONE
Low testosterone in men will result in a lowering of D1 activity without changing pituitary D2. Thus, a drop in testosterone will result in lower tissue levels of T3 without producing an elevation of TSH. Environmental factors, including pesticides, plastics, and other pollutants, have resulted in a significant decrease in the average testosterone levels for men, so most men will have, at least, a relative deficiency of testosterone. Major laboratories have, unfortunately, reduced the “normal” range of free testosterone to maintain the 95th percentile as normal, the result being that many abnormally low levels will now be considered normal.
In particular, the majority of male diabetics and those with insulin resistance will have suppressed testosterone level that is in the low or low-normal range, which further suppresses D1 and tissue T3 levels and perpetuates the weight gain or inability to lose weight – worsening these conditions.
GROWTH HORMONE
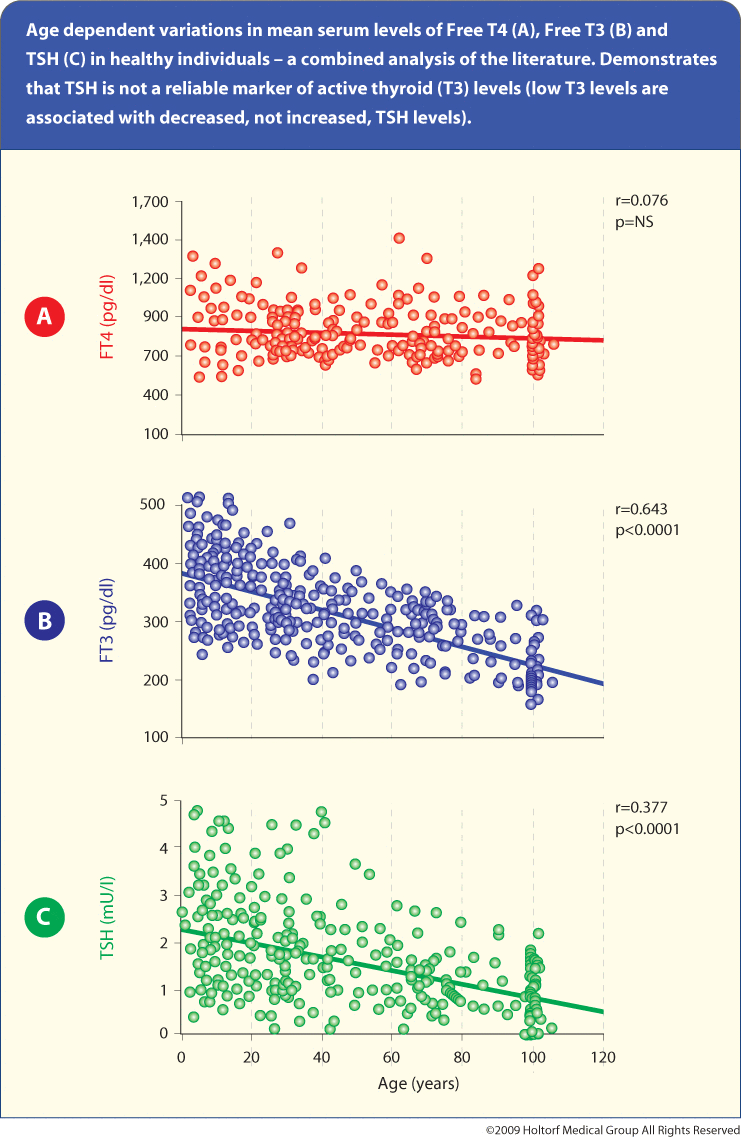
Growth hormone deficiency reduces T4 to T3 conversion and increases reverse T3 while supplementation with growth hormone improves T4 to T3 conversion and reduces reverse T3. The age-associated decline in growth hormone certainly contributes to the reduced T3 levels with age not detected by TSH and T4 testing (see Figure 5).fig4
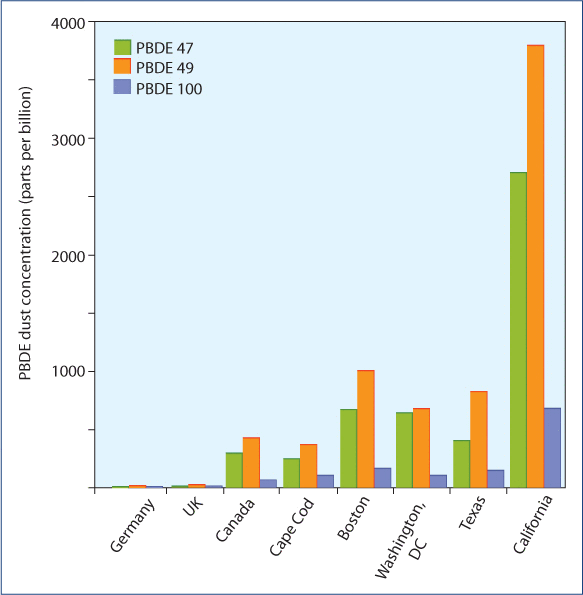
Reprinted (adapted) with permission from Zota AR, Rudel RA, Morello-Frosch RA, Brody JG. Elevated house dust and serum concentrations of PBDEs in California: unintended consequences of furniture flammability standards? Environ. Sci. Technol. 2008;42:8158–64.Copyright (2008) American Chemical Society.
 Schwartz E, Holtorf, K. Hormones in Wellness and Disease Prevention: Common Practices, Current State of the Evidence, and Questions for the Future. Prim Care Clin Office Pract. 2008;35:669–705.
Schwartz E, Holtorf, K. Hormones in Wellness and Disease Prevention: Common Practices, Current State of the Evidence, and Questions for the Future. Prim Care Clin Office Pract. 2008;35:669–705.
2. Mariotti S, Barbesino G, Caturegli P, et al. Complex alteration of thyroid function in healthy centenarians. J Clin Endocrinol Met. 1993;77(5):1132.
3. Verheecke, P. Free triiodothyronine concentration in serum of 1050 euthyroid children is inverselyrelated to their age. Clin Chem. 1997;43(6):963–7.
4. Quest Diagnostics assay validation free T3, free T4 and TSH, San Juan Capistrano, CA.
5. Jankowska H. The effect of age on parameters of thyroid function. Endokrynol Pol. 1993;44(2):117–27.
INDIVIDUAL VARIATIONS IN DEIODINASE
The relative amounts of D1, D2, and D3 vary in different tissues among different individuals and under varying conditions, resulting in hundreds of possible symptoms with hypothyroidism; some people have one symptom, some have a few, and some people have many, depending on the relative level of T3 in each tissue. Unfortunately, serum thyroid levels often do not accurately reflect intracellular tissue levels or levels in a particular tissue.
SUMMARY
With an improved understanding of thyroid physiology that includes the local control of intracellular activation and deactivation of thyroid hormones by deiodinases, it becomes clear that standard thyroid tests often do not reflect the thyroid status in the tissues of the body, other than the pituitary. This is especially true with physiologic and emotional stress, depression, dieting, obesity, leptin insulin resistance, diabetes, chronic fatigue syndrome and fibromyalgia, inflammation, autoimmune disease, or systemic illness. Consequently, it is inappropriate to rely on a normal or low TSH as an adequate or sensitive indicator of normal or low tissue levels of T3 in the presence of any such conditions, making the TSH a poor marker for the body’s overall thyroid level.
In order to be appropriately and thoroughly evaluated for thyroid dysfunction and obtain optimal treatment, it is important that patients find a thyroidologist who understands the limitations of standard thyroid testing and can clinically evaluate patients by taking an extensive inventory of potential signs and symptoms that may be due to low tissue thyroid levels despite normal standard thyroid tests.
DISCLOSURE OF INTERESTS
Dr. Holtorf supplies supplements from Holtraceuticals to support thyroid function to his own patients, outside the submitted work.
REFERENCES
- Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenode-iodinases.
- Silva JE, Larsen PR. Pituitary nuclear 3,5,3′-triiodothyronine and thyrotropin secretion: an explanation for the effect of thyroxine.
- Koenig RJ, Leonard JL, Senator D, Rappaport N. Regulation of thyroxine 5′-deiodinase activity by 3,5,3′-triiodothyronine in cultured rat anterior pituitary cells.
- Silva JE, Dick TE, Larsen PR. The contribution of local tissue thyroxine monodeiodination to the nuclear 3,5,3′-triiodothyroinine in pituitary, liver and kidney of euthyroid rats.
- Visser TJ, Kaplau MM, Leonard JL, Larsen PR. Evidence for two pathways of iodothyroinine 5′-deiodination in rat pituitary that differ I kinetics, propyl-thiouracil sensitivity, and response to hypothyroidism.
- Larsen PR, Silva JE, Kaplan MM. Relationship between circulation and intracellular thyroid haomrones: physiological and clinical implications.
- Kaplan MM. The role of thyroid hormone deiodination in the regulation of hypothalamo-pituitary function progress in neuroendocrinology.
- Peeters RP, Geyten SV, Wouters PJ, et al. Tissue thyroid hormone levels in critical illness.
- Peeters RP, Wouters PJ, Toor HV, et al. Serum 3,3′,5′-triiodothyronine (rT3) and 3,5,3′-triiodothyronine/rT3 are prognostic markers in critically ill patients and are associated with postmortem tissue deiodinase activities.
- Campos-Barros A, Hoell T, Musa A, Sampaolo S, et al. Phenolic and tyrosyl ring iodothyronine deiodination and thyroid hormone concentrations in the human central nervous system.
- Chopra IJ, Chopra U, Smith SR, et al. Reciprocal changes in serum concentrations of 3,3′,5-triiodothyronine (T3) in systemic illnesses.
- Chopra IJ, Williams DE, Orgiazzi J, Solomon DH. Opposite effects of dexamethasone on serum concentrations of 3,3′,5′- triiodothyronine (reverse T3) and 3,3′,5-triiodothyronine (T3).
- Duick DS, Warren DW, Nicoloff JT, Otis CL, Croxson MS. Effect of single dose dexamethasone on the concentration of serum triiodothyronine in man.
- Cavalieri RR, Castle JN, McMahon FA. Effects of dexamethasone on kinetics and distribution of triiodothyronine in the rat.
- Bianco AC, Nunes MT, Hell NS, Maciel RMB. The role of glucocorticoids in the stress-induced reduction of extrathyroidal 3,5,3′-triiodothyronine generation in rats.
- DeGroot LJ. Non-thyroidal illness syndrome is functional central hypothyroidism, and if severe, hormone replacement is appropriate in light of present knowledge.
- Reed HL, Brice D, Shakir KM, Burman KD, et al. Decreased free fraction of thyroid hormones after prolonged Antarctic residence.
- Forhead AJ, Curtis K, Kaptein E, Visser TJ, Fowden Al. Developmental control of iodothyronine deiodinases by cortisol in the ovine fetus and placenta near term.
- Nicoloff JT, Fisher DA, Appleman MD. The role of glucocorticoids in the regulation of thyroid function in man.
- Brabant G, Brabant A, Ranft U, Ocran K, et al. Circadian and pulsatile thyrotropin secretion in euthyroid man under the influence of thyroid hormone and glucocorticoid administration.
- Benker G, Raida M, Olbricht T, et al. TSH secretion in Cushing’s Syndrome: relation to glucocorticoid excess, diabetes, goitre, and the ‘Sick Euthyroid Syndrome’.
- Mebis L, Langouche L, Visser TJ, Van den Berghe G. The type II iodothyronine is up-regulated in skeletal muscle during prolonged critical illness.
- Linnoila M, Lamberg BA, Potter WZ, Gold PW, Goodwin FK. High reverse T3 levels in manic and unipolar depressed women.
- Kjellman BF, Ljunggren JG, Beck-Friis J, Wetterberg L. Reverse T3 levels in affective disorders.
- Jackson I. The thyroid axis and depression.
- Gitlin M, Altshuler LL, Frye MA, Suri M, Huynh EL, et al. Peripheral thyroid hormones and response to selective serotonin reuptake inhibitors.
- Clausen P, Mersebach H, Nielsen B, et al. Hypothyroidism is associated with signs of endothelial dysfunction despite 1-year replacement therapy with levothyroxine.
- Duval F, Mokrani MC, Bailey P, Correa H, et al. Thyroid axis activity and serotonin function major depressive episode.
- Unden F, Ljunggren JG, Kjellman BF, Beck-Friis J, Wetterberg L. Twenty-four-hour serum levels of T4 and T3 in relation to decreased TSH serum levels and decreased TSH response to TRH in affective disorders.
- Linnoila M, Lamberg BA, Rosberg G, Karonen SL, Welin MG. Thyroid hormones and TSH, prolactin and LH responses to repeated TRH and LRH injections in depressed patients.
- Kirkegaard C, Faber J. Altered serum levels of thyroxine, triiodothyronines and diiodothyronines in endogenous depression.
- Sintzel F, Mallaret M, Bougerol T. Potentializing of tricyclics and serotoninergics by thyroid hormones in resistant depressive disorders.
- Panicker V, Evans J, Bjoro T, Asvold BO. A paradoxical difference in relationship between anxiety, depression and thyroid function in subjects on and not on T4: findings from the Hunt study.
- Thompson FK. Is there a thyroid-cortisol-depression axis?
- Forman-Hoffman V, Philibert RA. Lower TSH and higher T4 levels are associated with current depressive syndrome in young adults.
- Cole DP, Thase ME, Mallinger AG, et al. Slower treatment response in biolar depression predicted by lower pretreatment thyroid function.
- Premachandra1 BN, Kabir MA, Williams IK. Low T3 syndrome in psychiatric depression.
- Isogawa K, Haruo Nagayama H, Tsutsumi T, et al. Simultaneous use of thyrotropin-releasing hormone test and combined dexamethasone/corticotropine-releasing hormone test for severity evaluation and outcome prediction in patients with major depressive disorder.
- Sullivan GM, Hatterer JA, Herbert J, Chen X, Rosse SP. Low levels of transthyretin in CSF of depressed patients.
- Hatterer JA, Herbert J, Hidaka C, Roose SP, Gorman JM. CSF transthyretin in patients with depression.
- Whybrow PC, Coppen A, Prange AJ, Noguera R, Bailey JE. Thyroid function and the response to liothyronine in depression.
- Kirkegaard C, Faber J. Free thyroxine and 3,3′,5′-triiodothyroidnine levels in cerebralspinal fluid in patetns with endogenous depression.
- Kirkegaard C. The thyrotropin response to thyrotropin-releasing hormone in endogenous depression.
- Baumgartner A, Graf KJ, Kurten I, Meinhold H. The hypothalamic-pituitary-thyroid axis in psychiatric patients and healthy subjects.
- Stipcevic T, Pivac N, Kozarie-Kovacic D, Muck-Seler D. Thyroid activity in patients with major depression.
- Cheron RG, Kaplan MM, Larsen PR. Physiological and pharmacological influences on thyroxine to 3,5,3′-triiodothyronine conversion and nuclear 3,5,3′-triiodthyroidne binding in rat anterior pituitary.
- Araujo RL, Andrade BM, da Silva ML, et al. Tissue-specific deiodinase regulation during food restriction and low replacement dose of leptin in rats.
- Leibel RL, Jirsch J. Diminished energy requirements in reduced-obese patients.
- Fontana L, Klein S, Holloszy JO, Premachandra BN. Effect of long-term calorie restriction with adequate protein and micronutrients on thyroid hormones.
- Croxson MS, Ibbertson HK. Low serum triiodothyronine (T3) and hypothyroidism.
- Silva JE, Larsen PR. Hormonal regulation of iodothyronine 5-deiodinase in rat brown adipose tissue.
- Krotkiewski M, Holm G, Shono N. Small doses of triiodothyronine can change some risk factors associated with abdominal obesity.
- Krotkiewski M. Thyroid hormones and treatment of obesity.
- Dagogo-Jack S. Human leptin regulation and promis in pharmacotherapy.
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, et al. Serum immunoreactive-leptin. Concentrations in normal-weight and obese humans.
- Dagogo-Jack S, Tanellis C, Paramore D, Brother SJ, Land TM. Plasma leptin and insulin relationships in obese and nonobese human.
- Maffei M, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob NAN in obese and weight-reduced subjects.
- Hassink SG, Sheslow DV, de Lancey E, Opentanova I, Considine RV, Caro JF. Serum leptin in children with obesity. Relationship to gender and development.
- Kozlowska L, Kozlowska L, Rosolowska-Huszcz. Leptin, thyrotropin, and thyroid hormones in obese/overweight women before and after two levels of energy deficit.
- Fekete C, et al. differential effects of central leptin, insulin, or glucose administration during fasting on the hypothalamic-pituitary-thyroid axis and feeding-related neurons in the arcuate nucleus.
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting.
- Legradi G, Emerson CH, Ahima RS, Flier JS, Lechan RM. Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus.
- Zimmermann-Belsing T, et al. Ciruclation leptin and thryoid dysfunction.
- Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG. Central nervous system control of food intake.
- Mantzoros CS, Moschos SJ. Leptin: in search of role(s) in human physiology and path physiology.
- Fruhbeck G, Jebb SA, Prentice AM. Leptin: physiology and pathophysiology.
- Flier JS, Harris M, Hollenber A. Leptin, nutrition and the thyroid: the why, the wherefore and the wiring.
- Gon DW, He Y, Karas M, Reitman M. Uncoupling protein-3 is a mediator of thermogenesis regulated by thyroid hormone, beta 3-adernergic agonists and leptin.
- Cusin I, Rouru J, Visser T, Burger AG, Rohner-Jeanrenaud F. Involvement of thyroid hormones in the effect of intracerebroventricular leptin infusion on un-coupling protein-3 expression in rat muscle.
- Rosenbaum M, Godmsith R, et al. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight.
- Rosenbaum M, Murphy, et al. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulation concentration of thyroid hormones.
- Leibel RL, et al. Changes in energy expenditure resulting from altered body weight.
- Rosenbaum M, et al. The effects of changes in body and thyroid function.
- Ahima R, et al. Role of leptin in the neuroendocrine response to fasting.
- Rosenbaum M, et al. Effects of weight change on plasma leptin concentrations and energy expenditure.
- Légrádi G, Emerson CH, Ahima RS, Flier JS, Lechan RM. Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonu-cleic acid in neurons of the hypothalamic paraventricular nucleus.
- Boozer CN, Leibel RL, Love RJ, Cha MC, Aronne LJ. Synergy of leptin and sibutramine in treatment of diet-induced obesity in rats.
- Campfield LA, et al. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks.
- Farooqi I, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency.
- Chehab F. Leptin as a regulator of adipose tissue mass and reproduction.
- Rosenbaum K, et al. The role of leptin in human physiology.
- Naslund E, et al. Associations of leptin, insulin resistance and thyroid function with long-term weight loss in dieting reduced-obese men.
- Doucette E, et al. Appetite after weight-loss by energy restriction and a low-fat diet-exercise follow up.
- Lisboa PC, Oliveira KJ, Cabanelas A, Ortiga-Carvalho TM, Pazos-Moura CC. Acute cold exposure, leptin, and somatostatin analog (octreotide) modulate thyroid 5′-deiodinase activity.
- Cabanelas A, Lisboa PC, Moura EG, Pazos-Moura CC. Leptin acute modulation of the 5′-deiodinase activities in hypothalamus, pituitary and brown adipose tissue of fed rats.
- Cettour-Rose P, Burger AG, Meier CA, Visser TJ, et al. Central stimulatory effect of leptin on T3 production is mediated by brown adipose tissue type II deiodinase.
- Fekete C, Kelly J, Mihaly E, Sarkar S, Rand WM, Legradi G, et al. Neuropeptide Y has a central inhibitory action on the hypothalamic–pituitary–thyroid axis.
- Fekete C, Legradi G, Mihaly E, Huang QH, Tatro JB, Rand WM, et al. A-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression.
- Legradi G, Emerson CH, Ahima RS, et al. Arcuate nucleus ablation prevents fasting-induced suppression of ProTRH mRNA in the hypothalamic paraventricular nucleus.
- Vignati L, Finley RJ, Hagg S, Aoki TT. Protein conservation during prolonged fast: a function of triiodothyronine levels.
- Katzeff HL, Selgrad C. Impaired peripheral thyroid hormone metabolism in genetic obesity.
- Islam S, Yesmine S, Khan SA, Alam NH, Islam S. A comparative study of thyroid hormone levels in diabetic and non-diabetic patients.
- Pittman CS, Suda AK, Chambers JB, McDaniel HG, Ray GY. Abnormalities of thyroid hormone turnover in patients with diabetes mellitus before and after insulin therapy.
- Saunders J, Hall SHE, Sonksen PH. Thyroid hormones in insulin requiring diabetes before and after treatment.
- Chamras H, Hershman JM. Effect of diabetes mellitus on thyrotropin release from isolated rat thyrotrophs.
- Ortiga-Carvalho TM, Curty FH, Nascimento-Saba CC, Moura EG, et al. Pituitary neuromedin B content in experimental fasting and diabetes mellitus and correlation with thyrotropin secretion.
- Jolin T, Gonzalez C. Thyroid iodine metabolism in streptozotocin-diabetic rats.
- Montoya E, Gonzalez C, Lamas L, Jolin T. Changes of the hypothalamus-pituitary-thyroid axis in streptozotocin-diabetic rats during adaptation to a low iodine diet.
- Pericas I, Jolin T. The effect of streptozotocin-induced diabetes on the pituitary-thyroid axis in goitrogen-treated rats.
- Docter R, Krenning EP, de Jong M, et al. The sick euthyroid syndrome: changes in thyroid hormone serum parameters and hormone metabolism.
- Chopra IJ. Euthyroid sick syndrome: is it a misnomer?
- Nagaya T, Fujieda M, Otsuka G, et al. A potential role of activated NF-Kappa B in the pathogenesis of euthyroid sick syndrome.
- Chopra IJ, Solomon DH, Hepner GW, et al. Misleadingly low free thyroxine index and usefulness of reverse triiodothyronine measurement in nonthyroidal illnesses.
- Van der Poll T, Romijn JA, Wiersinga WM, et al. Tumor necrosis factor: a putative mediator of the sick euthyroid syndrome in man.
- Stouthard JM, van der Poll T, Endert E, et al. Effects of acute and chronic interleukin-6 administration on thyroid hormone metabolism in humans.
- Corssmit EP, Heyligenberg R, Endert E, et al. Acute effects of interferon-alpha administration on thyroid hormone metabolism in healthy men.
- Zoccali C, Tripepi G, Cutrupi S, et al. Low triiodothyronine: a new facet of inflammation in end-stage renal disease.
- Chopra IJ, Sakane S, Teco GNC. A study of the serum concentration of tumor necrosis factor-alpha in thyroidal and nonthyroidal illnesses.
- Boelen A, Platvoet-Ter Schiphorst MC, Wiersinga WM. Association between serum interleukin-6 and serum 3,5,3′-triiodothyronine in nonthyroidal illness.
- Hashimoto H, Igarashi N, Yachie A, Miyawaki T, et al. The relationship between serum levels of interleukin-6 and thyroid hormone in children with acute respiratory infection.
- van der Poll T, Romijn JA, Wiersinga WM, Sauerwein HP. Tumor necrosis factor: a putative mediator of the sick euthyroid syndrome in man.
- Altomonte L, et al. Serum levels of interleukin-1alpha, tumor necrosis factor-alpha and interleukin-2 in rheumatoid arthritis. Correlation with disease activity.
- Espersen GT, et al. Tumor necrosis factor-alpha and interleukin-2 in plasma from rheumatoid arthritis patients in relation to disease.
- Lowe JC, Garrison RL, Reichman AJ, et al. Effectiveness and safety of T3 (triiodothyronine) therapy for euthyroid fibromyalgia: a double-blind placebo-controlled response-driven crossover study.
- Lowe JC, Reichman AJ, Yellin J. The process of change during T3 treatment for euthyroid fibromyalgia: a double-blind placebo-controlled crossover study.
- Lowe JC, Garrison RL, Reichman AJ, et al. Triiodothyronine (T3) treatment of euthyroid fibromyalgia: a small-n replication of a double-blind placebo-controlled crossover study.
- Yellin BA, Reichman AJ, Lowe JC. The process of change during T3 treatment for euthyroid fibromyalgia: a double-blind placebo-controlled crossover study.
- Neeck G, Riedel W. Thyroid function in patients with fibromyalgia syndrome.
- Morley JE. The endocrinology of the opiates and opioid peptides.
- Krulich L, Giachetti A, Marchlewska-Koj A, et al. On the role of central norandrenergic and dopaminergic systems in the regulation of TSH secretion in the rat.
- Lomax P, Kokka N, George R. Thyroid activity following intracerbral injection of morphine in the rat.
- Morley JE, Yamada T, Shulkes A, et al. Effects of morphine addiction and withdrawal on thyrotropin releasing hormone (TRH), somatostatin (SLI) and vasoactive intestinal peptide (VIP).
- Dons RF. Changes in Triiodothyronine mark severe pain syndrome: a case report.
- Watanabe C, Yoshida K, Kasanuma Y, Kun Y, Satoh H. In utero methylmercury exposure differentially affects the activities of selenoenzymes in the fetal mouse brain.
- Ellingsen DG, Efskind J, Haug E, Thomassen Y, Martinsen I, Gaarder PI. Effects of low mercury vapour exposure on the thyroid function in chloralkali workers.
- Moriyama K, Tagami T, Akamizu T, Usui T, et al. Thyroid hormone action is disrupted by bisphenol A as an antagonist.
- Zoeller RT, Bansal R, Parris C. Bisphenol-A. An environmental contaminant that acts as a thyroid hormone receptor antagonist in vitro, increases serum thyroxine, and alters RC3/neurogranin expression in the developing rat brain.
- Santini F, Mantovani A, Cristaudo A, et al. Thyroid function and exposure to styrene.
- Meeker JD, Calafat AM, Hauser R. Di(2-ethylhexyl) Phthalate metabolites may alter thyroid hormone levels in men.
- Massart F, Massai G, Placidi G, Saggese G. Child thyroid disruption by environmental chemicals.
- Heimeier RB, Buchholz DR, Shi YB. The xenoestrogen bisphenol A inhibits postembryonic vertebrate development by antagonizing gene regulation by thyroid hormone.
- Lema SC, Dickey JT, Schultz IR, Swanson P. Dietary exposure to 2,2′,4,4′-tetrabromodiphenyl ether (PBDE 47) alters thyroid status and thyroid hormone-regulated gene transcription in the pituitary and brain.
- De Groot Leslie J. Non-Thyrodial illness syndrome is a manifestation of hypothalamic-pituitary dysfunction, and in view of current evidence, should be treated with appropriate replacement therapies.
- Schilling JU, Zimmermann T, Albrecht S, et al. Low T3 syndrome in multiple trauma patients – a phenomenon or important pathogenetic factor.
- Schulte C, Reinhardt W, Beelen D, et al. Low T3-syndrome and nutritional status as prognostic factors in patients undergoing bone marrow transplantation.
- Girvent M, Maestro S, Hernandez R, et al. Euthyroid sick syndrome, associated endocrine abnormalities, and outcome in elderly patients undergoing emergency operation.
- Maldonado LS, Murata GH, Hershman JM, et al. Do thyroid function tests independently predict survival in the critically ill.
- Vaughan GM, Mason AD, McManus WF, et al. Alterations of mental status and thyroid hormones after thermal injury.
- De Marinis L, Mancini A, Masala R, et al. Evaluation of pituitary-thyroid axis response to acute myocardial infarction.
- Kantor MJ, Leef KH, Bartoshesky L, et al. Admission thyroid evaluation in very-low-birthweight infants: association with death and severe intraventricular hemorrhage.
- Miyashita K, Murakami M, Iriuchijima T, Takeuchi T, Mori M. Regulation of rat liver type 1 iodothyronine deiodinasemRNA levels by testosterone.
- Harris AR, Vagenakis AG, Braverman LE. Sex-related differences in outer ring monodeiodination of thyroxine and 3,3′,5′- triiodothyronine by rat liver homogenates.
- Visser TJ, Leonard JL, Kaplan MM, Larsen PR. Kinetic evidence suggesting two mechanisms for iodothyronine 5′-deiodination in rat cerebral cortex.
- Leonard JL. Dibutyryl cAMP induction of Type II 5. deiodinase activity in rat brain astrocytes in culture.
- Silva JE, Gordon MB, Crantz FR, Leonard JL, Larsen PR. Qualitative and quantitative differences in pathways of extrathyroidial triiodothyronine generation between euthyroid and hypothyroid rats.
- Larsen PR. Thyroid-pituitary interaction: feedback regulation of thyrotropin secretion by thyroid hormones.
- Schimmel M, Utiger RD. Thyroidal and peripheral production of thyroid hormones: review of recent finding and their clinical implications.
- Silva JE, Leonard JL, Crantz FR, Larsen PR. Evidence for two tissue specific pathways for in vivo thyroxine 5′ deiodination in the rat.
- Silva JE, Larsen PR. Contributions of plasma triiodothyronine and local thyroxine monodeiodination to triiodothyronine to nuclear triiodothyronine receptor saturation in pituitary, liver, and kidney of hypothyroid rats: further evidence relating saturation of pituitary nuclear triiodothyronine receptors and the acute inhibition of thyroid-stimulating hormone release.
- Bianco AC, Silva JE. Nuclear 3,5,3′-triiodothyronine (T3) in brown adipose tissue: receptor occupancy and sources of T3 as determined by in vivo techniques.
- van Doorn JD, Roelfsema F, van der Heide D. Contribution from local conversion of thyroxine to 3,5,3′-triiodothyronine to cellular 3,5,3′-triiodothyronine in several organs in hypothyroid rats at isotope equilibrium.
- van Doorn JD, van der Heide D, Roelfsema F. Sources and quantity of 3,5,3′-triiodothyronine in several tissues of the rat.
- van Doorn JD, Roelfsema F, van der Heide D. Concentrations of thyroxine and 3,5,3′-triiodothyronine at 34 different sites in euthyroid rats as determined by an isotopic equilibrium technique.
- Eales JG, McLeese JM, Holmes JA, Youson JH. Changes in intestinal and hepatic thyroid hormone deiodination during spontaneous metamorphosis of the sea lamprey, Petromyzon marinus.
- Croteau W, Davey JC, Galton VA, St. Germain DL. Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues.
- Gereben B, Bartha T, Tu HM, Harney JW, Rudas P, Larsen PR. Cloning and expression of the chicken type 2 iodothyronine 5′-deiodinase.
- Tu HM, Kim SW, Salvatore D, Bartha T, et al. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone.
- Leonard JL, Kaplan MM, Visser TJ, Silva JE, Larsen PR. Cerebral cortex responds rapidly to thyroid hormones.
- Burmeister LA, Pachucki J, St. Germain DL. Thyroid hormones inhibit type 2 iodothyronine deiodinase in the rat cerebral cortex by both pre- and posttranslational mechanisms.
- Salvatore D, Bartha T, Harney JW, Larsen PR. Molecular biological and biochemical characterization of the human type 2 selenodeiodinase.
- Hosoi Y, Murakami M, Mizuma H, Ogiwara T, et al. Expression and regulation of type II iodothyronine deiodinase in cultured human skeletal muscle cells.
- Riskind PN, Kolodny JM, Larsen PR. The regional hypothalamic distribution of type II 5′-monodeiodinase in euthyroid and hypothyroid rats.
- Guadano-Ferraz A, Obregon MJ, St. Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain.
- Berry MJ, Banu L, Larsen PR. Type I iodothyronine deiodinase is a selenocysteine-containing enzyme.
- Maia AL, Berry MJ, Sabbag R, Harney JW, Larsen PR. Structural and functional differences in the dio1 gene in mice with inherited type 1 deiodinase deficiency.
- Kaplan MM, Utiger RD. Iodothyronine metabolism in liver and kidney homogenates from hypothyroid and hyperthyroid rats.
- Harris ARC, Fang SL, Vagenakis AG, Braverman LE. Effect of starvation, nutrient replacement, and hypothyroidism on in vitro hepatic T4 to T3 conversion in the rat.
- Berry MJ, Kates AL, Larsen PR. Thyroid hormone regulates type I deiodinase messenger RNA in rat liver.
- Maia AL, Harney JW, Larsen PR. Pituitary cells respond to thyroid hormone by discrete, gene-specific pathways.
- Fliers E, Alkemade A, Wiersinga WM. The hypothalamic-pituitary-thyroid axis in critical illness.
- Danforth EJ, Desilets EJ, Jorton ES, Sims EAH, et al. Reiprocal serum triiodothryronine (T3) and reverse (rT3) induced by altering the carbohydrate content of the diet.
- Palmbald J, Levi J, Burger AG, Melade H, Westgren U, et al. Effects of total energy withdrawal (fasting) on the levels of growth hormone, thryrotropin, cortisol, noradrenaline, T4, T3 and rT3 in healthy males.
- De Jong F, den Heijer T, Visser TJ, et al. Thyroid hormones, dementia, and atrophy of the medical temporal lobe.
- Goichot B, Schlienger JL, Grunenberger F, et al. Thyroid hormone status and nutrient intake in the free-living elderly. Interest of reverse triiodothyronine assessment.
- Visser TJ, Lamberts WJ, Wilson JHP, Docter WR, Hennemann G. Serum thyroid hormone concentrations during prolonged reduction of dietary intake.
- Okamoto R, et al. Adverse effects of reverse triiodothyronine on cellular metabolism as assessed by 1H and 31P NMR spectroscopy.
- Tien ES, Matsui K, Moore R, Negishi M. The nuclear receptor constitutively active/androstane receptor regulates type 1 deiodinase and thyroid hormone activity in the regenerating mouse liver.
- Benvenga S, Cahnmann HJ, Robbins J. Characterization of thyroid hormone binding to apolipoprotein-E: localization of the binding site in the exon 3-coded domain.
- Sechman A, Niezgoda J, Sobocinski R. The relationship between basal metabolic rate (BMR) and concentrations of plasma thyroid hormones in fasting cockerels.
- Pittman JA, Tingley JO, Nickerson JF, Hill SR. Antimetabolic activity of 3,3′,5′-triiodo-dl-thyronine in man.
- Santini F, Chopra IJ, Hurd RE, Solomon DH, Teco GN. A study of the characteristics of the rat placental iodothyronine 5-monodeiodinase: evidence that is distinct from the rat hepatic iodothyronine 5-monodeiodinase.
- Silva JE, Leonard JL. Regulation of rat cerebrocortical and adenophypophyseal type II 5′-deidodinase by thyroxinem triiodothyronine, and reverse triiodothyronine.
- Obregon MJ, Larsen PR, Silva JE. The Role of 3,3′,5′-triiodothyroinine in the regulation of type II iodothyronin 5′-deiodinase in the rat cerebral cortex.
- Chopra IJ. A study of extrathyroidal conversion of thyroxine (T4) to 3,3′,5-triiodothyronine (T3) in vitro.
- Mitchell AM, Manley SW, Rowan KA, Mortimer RH. Uptake of reverse T3 in the human choriocarcinoma cell line, JAr.
- Van der Geyten S, Buys N, Sanders JP, Decuypere E, et al. Acute pretranslational regulation of type III iodothyronine deiodinase by growth hormone and dexamethasone in chicken embryos.
- Peeters RP, Wouters PJ, Kaptein E, et al. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients.
- Annewieke W, van den Beld AW, Visser TJ, Feelders RA, et al. Thyroid hormone concentrations, disease, physical function and mortality in elderly men.
- Brent GA, Hershman JM. Thyroxine therapy in patients with severe nonthyroidal illnesses and low serum thyroxine concentration.
- Escobar-Morreale HF, Obregon MJ, Escobar del Rey F, Morreale de Escobar G. Replacement therapy for hypothyroidism with thyroxine alone does not ensure euthyroidism in all tissues, as studied in thyroidectomized rats.
- Lomenick JP, El-Sayyid M, Smith WJ. Effect of levo-thyroxine treatment on weight and body mass index in children with acquired hypothyroidism.
- Acker CG, Singh AR, Flick RP, et al. A trial of thyroxine in acute renal failure.
- Samuels MH, Schuff KG, Carlson NE, Carello P, Janowsky JS. Health status, psychological symptoms, mood, and cognition in L-thyroxine-treated hypothyroid subjects.
- Cooke RG, Joffe RT, Levitt AJ. T3 augmentation of antidepressant treatment in T4-replaced thyroid patients.
- Bettendorf M, Schmidt KG, Grulich-Henn J, et al. Tri-idothyronine treatment in children after cardiac surgery: a double-blind, randomized, placebo-controlled study.
- Pingitore A, Galli E, Barison A, et al. Acute effects of triiodothryronine replacement therapy in patients with chronic heart failure and low-T3 syndrome: a randomized placebo-controlled study.
- Meyer T, Husch M, van den Berg E, et al. Treatment of dopamine-dependent shock with triiodothyronine: preliminary results.
- Dulchavsky SA, Hendrick SR, Dutta S. Pulmonary biophysical effects of triiodothyronine (T3) augmentation during sepsis induced hypothyroidism.
- Novitzsky D, Cooper DKC, Human PA, et al. Triiodothyronine therapy for heart donor and recepient.
- Dulchavsky SA, Maitra SR, Maurer J, et al. Beneficial effects of thyroid hormone administration in metabolic and hemodynamic function in hemorrhagic shock.
- Klemperer JD, Klein I, Gomez M, et al. Thyroid hormone treatment after coronary-artery bypass surgery.
- Gomberg-Maitland M. Thyroid hormone and cardiovascular disease.
- Dulchavsky SA, Kennedy PR, Geller ER, et al. T3 preserves respiratory function in sepsis.
- Novitzky D, Cooper DK, Reichart B. Hemodynamic and metabolic responses to hormonal therapy in brain-dead potential organ donors.
- Hamilton MA, Stevenson LW, Fonarow GC, et al. Safety and hemodynamic effects of intravenous triiodothyronine in advanced congestive heart failure.
- Klemperer JD, Klein IL, Ojamaa K, et al. Triidothyronine therapy lowers the incidence of atrial fibrillation after cardiac operations.
- Smidt-Ott UM, Ascheim DD. Thyroid hormone and heart failure.
- Lim VS, Passo C, Murata Y, Ferrari E, et al. Reduced triiiodthyronine content in liver but not pituitary of the uremic rat model: demonstration of changes compatible with thyroid hormone deficiency in liver only.
- LoPresti JS, Eigen A, Kaptein E, Anderson KP, et al. Alterations in 3,3′5′-triiodothyronine metabolism in response to propylthiouracil, dexamethasone, and thyroxine administration in man.
- Cremaschi GA, Gorelik G, Klecha AJ, Lysionek AE, Genaro AM. Chronic stress influences the immune system through the thyroid axis.
- Burr WA, Ramsden DB, Griffiths RS, Black EG, Hoffenberg R, et al. Effect of a single dose of dexamethasone on serum concentrations of thyroid hormones.
- Saranteas T, Tachmintzis A, Katsikeris N, Lykoudis E, Mourouzis I, et al. Perioperative thyroid hormone kinetics in patients undergoing major oral and maxillo-facial operations.
- Joffe RT. A perspective on the thyroid and depression.
- Posternak M, Novak S, Stern R, Hennessey J, Joffe R, et al. A pilot effectiveness study: placebo-controlled trial of adjunctive L-triiodothyronine (T3) used to accelerate and potentiate the antidepressant response.
- Wekking EM, Appelhof BC, Fliers E, Schene AH, et al. Cognitive functioning and well-being in euthyroid patients on thyroxine replacement therapy for primary hypothyroidism.
- Escobar-Morreale HF, Escobar del Rey F, Obregon MJ, Morreale de Escobar G. Only the combined treatment with thyroxine and triiodothyroidine ensures euthyroidism in all tissues of the thyroidectomized rat.
- Sawka AM, Gerstein HC, Marriott MJ, MacQueen GM, Joffe RT. Does a combination regimen of thyroxine (T4) and 3,5,3′-triiodothyronine improved depressive symptoms better than T4 alone in patients with hypothyroidism? Results of a double-blind, randomized, controlled trial.
- Cooper-Kazaz R, Apter JT, Cohen R, Karagichev L, et al. Combined treatment with sertraline and liothyronine in major depression.
- Kelly T, Lieberman DZ. The use of triiodothyronine as an augmentation agent in treatment resistant bipolar II and bipolar disorder NOS.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: A STAR*D Report.
- Tennant F. Hormone treatments in chronic and intractable pain.
- Dore C, Hesp R, Wilkins D, et al. Prediction of energy requirements of obese patients after massive weight loss.
- Drenick EJ, Dennin HF. Energy expenditure in fasting obese men.
- Tulp OL, Mckee TD. Thiiodothyronine (T3) neogenesis in lean and obese LA/N-cp rats.
- Silva JE, Larsen PR. Interrelationships among thyroxine, growth hormone, and the sympathetic nervous system in the regulation of 5-iodothyronine deiodinase in rat brown adipose tissue.
- Loucks AB, Heath EM. Induction of low-T3 syndrome in exercising women occurs at the threshold of energy availability.
- Opstad PK, Falch D, Oktedalen O, Fonnum F, Wergeland R. The thyroid function in young men during prolonged exercise and the effect of energy and sleep deprivation.
- Dillman E, Gale C, Green W, et al. Hypothermia in iron deficiency due to altered triiodithyroidine metabolism.
- Smith SM, Johnson PE, Lukaski HC. In vitro hepatic thyroid hormone deiodination in iron-deficient rats: effect of dietary fat.
- Eftekhari MH, Keshavarz SA, Jalali M. The relationship between iron status and thyroid hormone concentration in iron-deficient adolescent Iranian girls.
- Zimmermann MB, Köhrle J. the impact of iron and selenium deficiencies on iodine and thyroid metabolism: biochemistry and relevance to public health.
- Beard J, Tobin B, Green W. Evidence for thyroid hormone deficiency in iron-deficient anemic rats.
- Zhou D, Kusnecov AW, Shurin MR, DePaoli M, Rabin BS. Exposure to physical and psychological stressors elevates plasma interleukin-6: relationship to the activation of the hypothalamic–pituitary–adrenal axis.
- Brunner EJ, Marmot MG, Nanchahal K, et al. Social inequality in coronary risk: central obesity and the metabolic syndrome. Evidence from the Whitehall II Study.
- Miller GE, Blackwell E. Turning up the heat: inflammation as a mechanism linking chronic stress, depression, and heart disease.
- Ranjit N, Diez-Roux AV, Shea S, et al. Psychosocial factors and inflammation in the Multi-Ethnic Study of Atherosclerosis.
- Davis MC, Zautra AJ, Younger J, Motivala SJ, et al. Chronic stress and regulation of cellular markers of inflammation in rheumatoid arthritis: implications for fatigue.
- Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link?
- Tilg H, Moschen AR. Insulin resistance, inflammation, and non-alcoholic fatty liver disease.
- Mohamed-Ali V, Goodrick S, Rawesh A, et al. Human subcutaneous adipose tissue secretes interleukin-6 but not TNF-a in vivo.
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor- a in human obesity and insulin resistance.
- Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid.
- Liu S, Tinker L, Song Y, Rifai N, et al. A prospective study of inflammatory cytokines and diabetes mellitus in a multiethnic cohort of postmenopausal women.
- Leonard BE. Inflammation, depression and dementia: are they connected?
- Maes M. Evidence for an immune response in major depression: a review and hypothesis.
- Maes M, Scharpé S, Meltzer HY, Bosmans E, et al. Relationships between interleukin-6 activity, acute phase proteins, and function of the hypothalamic-pituitary-adrenal axis in severe depression.
- Pfeilschifter J, Koditz R, Pfohl M, Schatz H. Changes in proinflamatory cytokine activity after menopause.
- Alexander RW. Inflammation and coronary artery disease.
- MRFIT Research Group, Kuller LH, Tracy RP, Shaten J, Meilahn EN. Relation of C-reactive protein and coronary heart disease in the MRFIT nested case-control study.
- Benvenuto R, et al. Tumor necrosis factor-alpha and interferon-alpha synthesis by cerebrospinal fluid-derived T cell clones in multiple sclerosis.
- Cohen MC, Cohen S. Cytokine function: a study in biologic diversity.
- Khan G. Epstein-Barr virus, cytokines, and inflammation: a cocktail for the pathogenesis of Hodgkin’s lymphoma?
- Takeshita S, et al. Induction of IL-6 and IL-10 production by recombinant HIV-1 envelope glycoprotein 41 (gp41) in the THP-1 human monocytic cell line.
- Coussens LM, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis.
- Cleeland CS, Bennett GJ, Dantzer R, et al. Are the symptoms of cancer and cancer treatment due to a shared biologic mechanism. A cytokine-immunologic model of cancer symptoms.
- Lee BN, Dantzer R, Langley KE, et al. A cytokine-based neuroimmunologic mechanism of cancer related symptoms.
- Malyszko J, Malyszko JS, Pawlak K, Mysliwiec M. Thyroid function, endothelium, and inflammation in hemodialyzed patients: possible relations?
- Boelen A, Kwakkel J, Alkemade A, Renckens R, et al. Induction of type 3 deiodinase activity in inflammatory cells of mice with chronic local inflammation.
- Takser L, Mergler D, Baldwin M, de Grosbois S, et al. Thyroid hormones in pregnancy in relation to environmental exposure to organochlorine compounds and mercury.
- Hesch RD, Brunner G, Soling HD. Conversion of thyroxine (T4) and triiodothyronine (T3) and the subcellular localization of the converting enzyme.
- Visser TJ, van der Does-Tobe I, Docter R, Hennemann G. Conversion of thyroxine into triiodothyronine by rat liver homogenate.
- Lakind JS, Naiman DQ. Biphenol A (BPA) daily intakes in the United States: estimates form the 2003–2004 NHANES urninary BPA data.
- Cone M. Californians have world’s highest levels of flame retardants.
- Travison TG, Araujo AB, O’Donnell AB, et al. A Population-level decline in serum testosterone levels in American men.
- Kupelian V, Hayes FJ, Link CL, et al. Inverse association of testosterone and the metabolic syndrome in men is consistent across race and ethnic groups.
- Kapoor D, Aldred H, Clark S, Channer KS, Jones TH. Clinical and biochemical assessment of hypogonadism in men with type 2 diabetes: correlations with bioavailable testosterone and visceral adiposity.
- Makhsida N, Shah J, Yan G. Hypogonadism and metabolic syndrome: implications for testosterone therapy.
- Jorgensen JOL, Pedersen SA, Laurberg P, Weeke J, et al. Effects of growth hormone therapy on thyroid function of growth hormone-deficient adults with and without concomitant thyroxine-substituted central hypothyroidism.
- Darras VM, Berghman LR, Vanderpooten A, Kuhn ER. Growth hormone acutely decreases type III deiodinase in chicken liver.
- De Jong FJ, Peeters RP, Jeijer TD, van der Deure WM, Hofman A, et al. The association of pohymorphism in the type 1 and 2 deiodinase genes with circulation thyroid hormone parameters and atrophy of the medial Temporal lobe.
- Apfelbaum M, Bostsarron J, Lacatis D. Effects of caloric restriction and excessive caloric intake on energy expenditure.
- Reinecker HC, et al. Enhanced secretion of tumor necrosis factor, IL-6, and IL-1 by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease.
- Gross V, et al. Inflammatory mediators in chronic inflammatory bowel disease.