Abstract
New research is demonstrating that thyroid hormone transport across cellular membranes plays an important role in intracellular triiodothyronine (T3) levels of peripheral and pituitary tissues and is proving to have considerable clinical significance. Reduced T4 and T3 transport into the cells in peripheral tissues is seen with a wide range of common conditions, including insulin resistance, diabetes, depression, bipolar disorder, hyperlipidemia, chronic fatigue syndrome, fibromyalgia, neurodegenerative diseases, migraines, stress, anxiety, chronic dieting and aging, while the intracellular T3 level in the pituitary often remains unaffected. The pituitary has different transporters than every other tissue in the body. The thyroid transporters in the body are very energy dependent and are affected by numerous conditions, including low energy states, toxins and mitochondrial dysfunction, while the pituitary remains unaffected. Because the pituitary remains largely unaffected and is able to maintain intracellular T3 levels while the rest of the body suffers from significantly reduced intracellular T3 levels, there is no elevation in thyroid-stimulating hormone (TSH) despite the presence of wide-spread tissue hypothyroidism, making the TSH and other standard blood tests a poor marker to determine the presence or absence of hypothyroidism. Because the T4 transporter is more energy dependent than the transporter for T3, it is also not surprising that T4 preparations are generally ineffective in the presence of such conditions, while T3 replacement is shown to be beneficial. Thus, if a patient with a normal TSH presents with signs or symptoms consistent with hypothyroidism, which may include low basal body temperature, fatigue, weight gain, depression, cold extremities, muscle aches, headaches, decreased libido, weakness, cold intolerance, water retention, slow reflex relaxation phase or PMS, a combination of both clinical and laboratory assessment, which may include a T3/reverse T3 ratio and the level of sex hormone binding globulin (SHBG), should be used to determine the likely overall thyroid status and if a therapeutic trail of straight T3 or a T4/T3 combination is indicated and not based solely on standard thyroid function tests.
INTRODUCTION
New research is demonstrating that thyroid hormone transport across cellular membranes plays an important role in tissue thyroid levels and is proving to have significant clinical significance. Thus, physicians who evaluate or treat thyroid dysfunction would benefit from an appreciation of this topic. Unfortunately, many physicians are not aware of new developments in the current understanding of thyroid hormone transport, which has resulted in an overreliance on the sole use of standard thyroid blood tests (e.g., thyroid-stimulating hormone (TSH) and T4 levels) to determine the presence or absence of hypothyroidism. This has resulted in the misdiagnosis of many hypothyroid patients as being euthyroid. This overreliance on standard thyroid blood tests and the overconfidence of the diagnostic accuracy of standard thyroid blood tests may be particularly troublesome in the presence of a wide range of common comorbid conditions present in a large percentage of the population.
Serum thyroid levels are, of course, commonly used as an indication of tissue thyroid activity. However, in order to have biological activity, T4 and T3 must cross the cellular membrane from the serum into the target cells. It follows that the activity of these transport processes may have a significant influence on the regulation of biological activity of these hormones. For about two and half decades it has been assumed that the rate and extent of uptake of thyroid hormone into the cells occurs by simple diffusion, which is driven by the concentration gradient of the free hormones in the serum. This “free hormone” or “diffusion hypothesis” was formulated in 1960 and assumes that the serum concentration of free thyroid hormones (free T4 and free T3) will ultimately determine intracellular thyroid hormone concentrations.
However, more recent data has challenged this hypothesized “diffusion” mechanism, showing that the transport of T4 and T3 across the cellular membrane into the cell requires active transport rather than passive diffusion. Despite accumulating evidence to the contrary, this misconceived “diffusion hypothesis” continues to be perpetuated among both primary physicians and endocrinologist.
CONDITIONS ASSOCIATED WITH ABNORMAL THYROID TRANSPORT
Since the transport of thyroid hormones into the cell is largely energy dependent, any condition associated with reduced production of the cellular energy (i.e., mitochondrial dysfunction) could also be associated with reduced transport of thyroid hormone into the cell. This can result in cellular hypothyroidism despite having standard blood tests in the “normal” range. Conditions linked with reduced mitochondrial function and impaired thyroid transport include: insulin resistance, diabetes and obesity; chronic and acute dieting; depression; anxiety; bipolar depression; neurodegenerative diseases; aging; chronic fatigue syndrome; fibromyalgia; migraines; chronic infections; physiologic stress and anxiety; cardiovascular disease; inflammation and chronic illness; and those with high cholesterol and triglyceride levels. Thus, standard blood tests can be very unreliable if any of these commonly occurring conditions are present.
It has also been shown that there are a number of substances that are produced by the body in response to dieting and physiologic stress that can negatively affect thyroid hormone transport. For instance, studies have shown that cell cultures incubated with serum from physiologically stressed or dieting individuals exhibit a dramatic reduction in the uptake of T4 into cells. This reduction correlated with the degree of stress.
It has also been demonstrated that there are a variety of distinct and specific transporters that are necessary for the transport of T4 and T3 into the cell. Furthermore, the transporter for T4 is much more energy dependent (it requires more energy) than the transporter for T3 (see Figure 1). Even slight reductions in cellular energy (i.e., mitochondrial function) can result in dramatic declines in the cellular uptake of T4, while the uptake of T3 appears to be much less affected. Therefore, the conditions listed above are particularly linked to an impaired transport of T4, resulting in cellular hypothyroidism. When there is reduced transport of T4 or T3 into the cells, the serum T4 and T3 levels increase (less is transported out of the serum). Thus, even though the production of T4 and T3 is diminished in chronic illness, the reduced transport into the cell will tend to raise serum levels, making the serum T4 and T3 remain normal or high-normal (see Figure 2). This is one of the primary reasons that cellular hypothyroidism is not routinely detected by standard serum T4 or T3 levels. A standard TSH level will also not detect such cellular hypothyroidism because the pituitary has completely different transporters that are not energy dependent. Therefore, while the rest of body has impaired thyroid transport, the pituitary experiences increased transport activity. Another reason that the standard TSH level may not detect cellular hypothyroidism is that the nuclear thyroid receptors in the pituitary (TR-B2) are different from those in other tissues (TR-a1 and TR-B1). Pituitary receptors can become saturated and turn off TSH while other tissues are lacking thyroid stimulation.
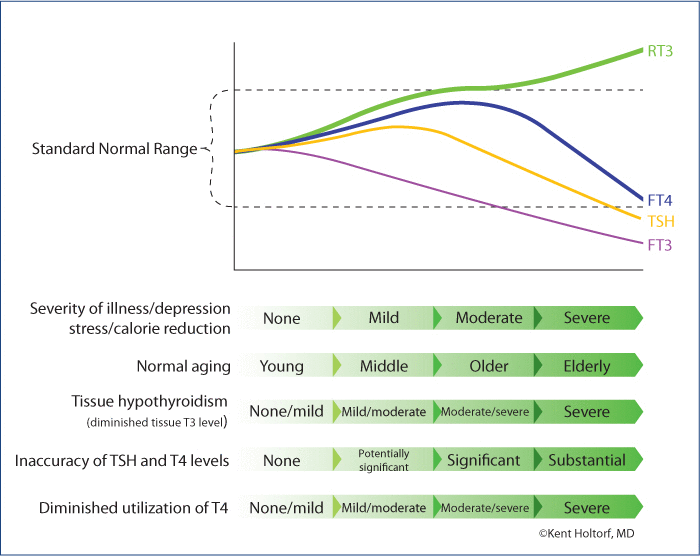
Serum thyroid levels with increasing physiologic stress and aging.
Associated serum thyroid levels with progressively decreasing tissue thyroid levels due to stress, illness, depression, calorie reduction or aging (Why standard blood tests lack sensitivity to detect low thyroid in the presence of such conditions).
Demonstrates why TSH levels lack the accuracy to detect cellular levels and the free T3/reverse T3 ratio is the most accurate method to determine cellular thyroid levels in the presence of physiologic stress, illness, depression or obesity.
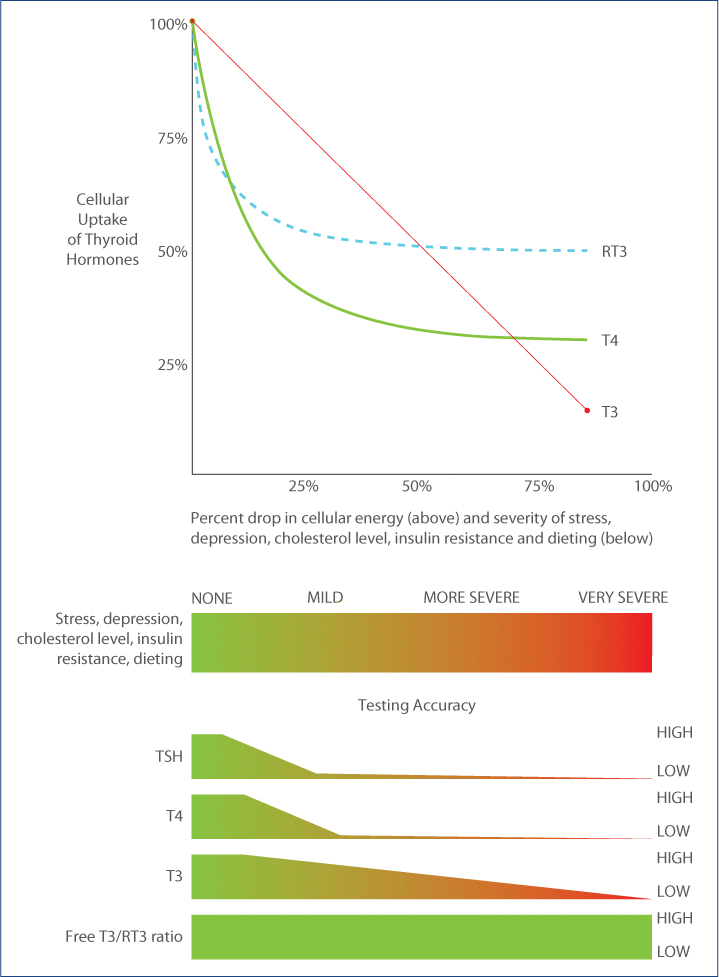
Thyroid transport of cellular energy. Why TSH testing is inaccurate and free T3/RT3 ratio is the best marker for thyroid transport.
Cellular thyroid levels do not correlate with serum levels if uptake into the cells is hindered. This occurs with chronic stress, depression, chronic dieting, diabetes, insulin resistance (obesity) or high cholesterol levels. Thus, with such conditions, TSH, T4 and T3 levels are not accurate measures of intracellular thyroid levels and cannot be used as reliable markers to determine the need for thyroid hormone supplementation. T4 uptake (utilization) drops much faster than T3 utilization as severity increases, making T4 replacement inappropriate for such conditions. Reverse T3 mirrors T4 uptake so high or high-normal reverse T3 is a marker for reduced uptake of T4 into the cell (and to a lesser extent T3) showing that there is a reduced overall tissue thyroid level requiring T3 supplementation (not T4). Utilizing the free T3/reverse T3 ratio does not suffer from the inaccuracies of standard tests and most closely correlates with cellular thyroid levels.
PITUITARY THYROID TRANSPORT DETERMINES TSH LEVELS
The pituitary is different than every cell in the body with its own distinct deiodinases, thyroid transporters, and high affinity thyroid receptors. As mentioned previously, the pituitary thyroid hormone transporters are not energy dependent and can thus maintain or increase the cellular uptake of T4 and T3 even in low energy states. This stands in stark contrast with transporters found in other parts of the body that would normally experience significantly reduced transport under similar circumstances.
One reason that pituitary thyroid hormone uptake is so resilient may be due to the fact that its T4 and T3 transporters are resistant to various environmental toxins and other substances produced by the body in response to physiologic stress and calorie reduction. Since thyroid hormone uptake in the pituitary is relatively unaffected by outside factors, the presence of intracellular hypothyroidism is not reflected by TSH testing, which is why TSH is a poor marker for cellular thyroid function in any tissue other than the pituitary. Benzodiazepine medications such as diazepam (Valium®), lorazapam (Atavan®), and alprazolam (Xanax®) are further examples where a given compound can inhibit T3 uptake into the cells of the body, but have no effect on transport of T3 into the pituitary.
St Germain et al. investigated the comparison between thyroid hormone transport into the pituitary versus the cells of the rest of the body. The authors demonstrated that the pituitary does not respond to calorie restriction (i.e., dieting) in the same way as the rest of the body. The dramatically reduced serum T4 and T3 levels that accompany dieting are associated with an increase in pituitary T3 receptor saturation (i.e., percentage of activated T3 receptors), which results in a decrease in TSH even when serum thyroid hormone levels were reduced by 50%. Wassen et al. also demonstrated the absence of inhibitory effects of chronic caloric deprivation and bilirubin on thyroid hormone uptake by pituitary cells compared to the reduced thyroid transport that occurred in the peripheral tissue (liver).
STRESS
Chronic emotional or physiologic stress can cause a significant reduction of transport of T4 into the cells of the body. For example, Sarne et al. added serum from different groups of individuals to cell cultures and measured the amount of T4 uptake from the serum into the cell. Their results showed that the serum from those with significant physiologic stress inhibited the uptake (transport) of T4 into the cell while the serum from the non-physiological stressed had no effect. These results demonstrate that serum T4 levels can be artificially elevated among physiologically stressed individuals, and thus serum T4 and TSH levels are poor markers for tissue thyroid levels in this patient population (see Figure 2). Substances produced by physiologic stress or calorie reduction (e.g., 3-carboxy-4-methyl-5-propyl-2-furan propanoic acid (CMPF), indoxyl sulfate, bilirubin and fatty acids), have been shown to reduce the cellular uptake of T4 by up to 42%, while having no effect on T4 or T3 uptake into the pituitary.
In addition to the above, numerous other studies have linked physiologic stress to reduced cellular uptake of T4 and T3. For instance, Arem et al. found that significant physiological stress was associated with dramatically reduced tissue levels of T4 and T3 (up to 79%) without a corresponding increase in TSH. The authors also found there was tissue variability in the level of suppression in different tissues, resulting in a significant variation when comparing the T4 and T3 levels in different tissues. This large variation of T4 and T3 levels in different tissues may explain the wide range and variation in individual symptoms of hypothyroidism.
DIETING
In a highly controlled study, Brownell et al. found that after repeated cycles of dieting, weight loss occurred at half the rate and weight gain occurred at three times the rate compared to controls with the same calorie intake. Furthermore, severe caloric restriction and weight cycling is shown to be associated with reduced cellular T4 uptake of 25%–50%. Therefore, successful weight loss is doomed to failure unless the reduced intracellular thyroid levels are addressed, but, as stated previously, this reduced cellular thyroid level is generally not detected by standard laboratory testing.
In a study published in the American Journal of Physiology-Endocrinology and Metabolism, Van der Heyden et al. studied the effect of calorie restriction (dieting) on the transport of T4 and T3 into the cell. They found that obese individuals in the processes of dieting exhibited a 50% reduction of T4 into the cell and a 25% reduction of T3 into the cell. This is thought to be due to the reduced cellular energy stores as well as increased levels of free fatty acids and non-esterified fatty acids (NEFA) in the serum. This data would help explain why standard thyroid blood tests are not accurate indicators of intracellular thyroid levels. This also explains why it is difficult for obese patients to lose weight; since, as calories are decreased, thyroid utilization is reduced and metabolism drops. Among patients with this type of thyroid hormone transport dysfunction (resulting in intracellular hypothyroidism) assessing the free T3/reverse T3 ratio can aid in a proper diagnosis, with a free T3/reverse T3 ratio of less than 0.2 being a marker for tissue hypothyroidism (when the free T3 is expressed in pg/mL (2.3–4.2 pg/mL) and the reverse T3 is expressed in ng/dL (8–25 ng/dL)) (see Figure 2).
REVERSE T3 (RT3)
TSH and serum T4 levels do not correlate well with intracellular thyroid levels. There are competing factors attributing to the serum free T3 levels; reduced T4 to T3 conversion will tend to reduce serum T3 levels while the reduced uptake into the cell tends to increase serum T3 (the T3 transporter is less affected by the low cellular energy than the T4 transporter). Increased rT3 levels are shown to be predominantly due to reduced transport into the cell and not due to increased T4 to rT3 conversion. Because the rT3 and T4 transporters are equally energy dependent, a high serum rT3 is shown to be a marker for reduced uptake of T4 into the cell.
Thus, rT3 is an excellent marker for identifying reduced cellular T4 and T3 levels that would not normally be detected by TSH or serum T4 and T3 tests. As a result, any increase (high or high-normal) of rT3 is not only an indicator of tissue hypothyroidism but also suggests that T4-only replacement would not be considered optimal therapy. While a high rT3 can occasionally be associated with hyperthyroidism, as the body tries to reduce cellular thyroid levels, this can be differentiated on the basis of symptoms and by utilizing the free T3/rT3 ratio, which correlates with intracellular thyroid levels (see Figure 2).
TREATMENT WITH T4
Levothyroxine (T4)-only replacement with products such as Synthroid® and Levoxyl® are the most widely accepted forms of thyroid replacement. This is based on a widely held assumption that the body will convert what it needs to the biologically active form (T3). Based on this assumption, many physicians and endocrinologists believe that the normalization of TSH with a T4 preparation demonstrates adequate tissue levels of thyroid, perpetuated by practice guidelines indicating T4 monotherapy as the standard of care. This assumption, however, had never been directly tested until two studies were published.
The first study investigated whether or not giving T4-only preparations will provide adequate T3 levels in varying tissues. Plasma TSH, T4, and T3 levels and 10 different tissue levels of T4 and T3 were measured after the infusion of 12–13 days of thyroxine. The authors found that the normalization of plasma TSH and T4 levels with T4-only preparations resulted in adequate tissue T3 levels in only a few select tissues types, namely brown adipose tissue, cerebellum and cortex. Almost every other tissue was found to be deficient. This data suggest that the use of T4 therapy (even if given at supra-physiological levels) cannot reasonably achieve normal tissue levels of T3. The authors conclude: “It is evident that neither plasma T4 nor plasma T3 alone permit the prediction of the degree of change in T4 and T3 concentrations in tissues… the current replacement therapy of hypothyroidism [giving T4] should no longer be considered adequate….”
The second study utilizing an experimental model compared plasma TSH, T4 and T3 levels and 13 different tissue levels of T4 and T3 when T4 or T4/T3 treatments were utilized. This study found that a combination of T4/T3 is required to normalize tissue levels of T3, and that the pituitary was able to maintain normal levels of T3 despite the rest of the body being hypothyroid on T4-only preparations. Under normal conditions it was shown that the pituitary will have 7 to 60 times the concentration of T3 when compared to other tissues of the body; and when thyroid levels drop, the pituitary was shown to have 40 to 650 times the concentration of T3 in other tissues. Thus, the pituitary is unique in its ability to concentrate T3 in the presence of diminished serum thyroid levels. Consequently, the pituitary levels of T3 and the subsequent TSH level are poor measures of tissue hypothyroidism, as almost the entire body can be severely hypothyroid despite having a normal TSH level.
The dramatic reduction of T4 cellular uptake that occurs with a wide variety of conditions also explains why T4 preparations are often associated with poor clinical response (i.e., continued residual symptoms). As stated by Hennemann et al. in Endocrine Reviews: “Even a small decrease in cellular ATP concentration results in a major reduction in the transport of T4 (and rT3) but only slightly affects T3 uptake.” This makes it inappropriate to use T4-only preparations when treating any condition associated with reduced mitochondrial function or ATP production, Thus, it is not surprising that T3 has been shown to be superior in such patient populations.
Fraser et al. investigated the correlation between tissue thyroid activity and serum blood tests (TSH, free T4, and T3) and published their results in theBritish Medical Journal. The authors concluded that “The serum concentration of thyroid stimulation hormone is unsatisfactory as the thyrotrophs in the anterior pituitary are more sensitive to changes in the concentration of thyroxin in the circulation than other tissues, which rely more on triiodothyronine (T3).” They found a suppressed or undetectable TSH was not an indication or a reliable marker of over replacement or hyperthyroidism. They state “It is clear that serum thyroid hormone and thyroid stimulating hormone concentrations cannot be used with any degree of confidence to classify patients as receiving satisfactory, insufficient, or excessive amounts of thyroxine replacement… The poor diagnostic sensitivity and high false positive rates associated with such measurements render them virtually useless in clinical practice… Further adjustments to the dose should be made according to the patient’s clinical response.”
Similar results were observed by Meier et al., who investigated the correlation of TSH and tissue thyroid effect. The authors concluded that “TSH is a poor measure for estimating the clinical and metabolic severity of primary overt thyroid failure... We found no correlations between the different parameters of target tissues and serum TSH.” They stated that signs and symptoms of thyroid effect and not the TSH should be used to determine the proper replacement dose.
Alevizaki et al. also studied the accuracy of using TSH to determine the proper T4 replacement dose, and found that the use of TSH tests (although common) cannot accurately detect cellular hypothyroidism in the majority of tissues, except for the pituitary. They conclude, “TSH levels used to monitor substitution, mostly regulated by intracellular T3 in the pituitary, may not be such a good indicator of adequate thyroid hormone action in all tissues.”
Likewise, Zulewski et al. found TSH to be an unsuitable measure of optimal or proper thyroid replacement, as they observed no correlation between TSH and tissue thyroid levels. However, serum T4 and T3 levels had some correlation, with T3 being a better indictor than T4. Based on their data, the authors concluded, “The ultimate test of whether a patient is experiencing the effects of too much or too little thyroid hormone is not the measurement of hormone concentration in the blood but the effect of thyroid hormones on the peripheral tissues [symptoms] .”
In fact, the positive predictive value of TSH (the likelihood that a suppressed TSH indicates over-replacement or hyperthyroidism) has been determined to be 16%. In other words, a suppressed TSH is not associated with hyperthyroidism or over-replacement 84% of the time, making it an inaccurate marker for determining an appropriate thyroid hormone replacement dose.
Additionally, TSH levels become an even worse indicator for an optimal replacement dose in the presence of the following comorbidities: insulin resistance or obesity; chronic calorie reduction; depression; bipolar depression; neurodegenerative diseases; advanced in age; chronic fatigue syndrome; fibromyalgia; migraines; chronic infections; stress or anxiety; heart failure or cardiovascular disease; inflammation or chronic illness; high cholesterol or triglyceride levels.
SHBG
Sex hormone binding globulin (SHBG) is formed in the liver in response to the tissue level of estrogen and thyroid. Thus, if a patient’s estrogen levels are considered to be adequate (natural or replaced), SHBG can be used as a marker for the T3 level in peripheral tissues. As expected, the SHBG is typically low in individuals with obesity, diabetes and insulin resistance due to the reduced transport of thyroid hormones into the cells with these individuals. Women with adequate estrogen levels should have an SHBG above 70 nmol/L and men above 25 nmol/L. If not, the diagnosis of low tissue levels of thyroid should be considered. The SHBG is more useful in women than men because the normal reference range is less broad in men, making it less sensitive and less clinically useful.
In addition to being useful in diagnosing low thyroid in patients, SHBG can also be used to determine the optimal replacement dose or preparation (T4, T4/T3 vs straight T3). Because thyroid replacement is given orally, it must first undergo first pass metabolism in the liver, resulting in significantly higher hepatic levels than in other peripheral tissues. Consequentially, if the SHBG is below 70 nmol/L in women or 25 nmol/L in men who are on thyroid replacement, the rest of body would not be expected to have adequate thyroid levels. Additionally, if the SHBG does not significantly increase with replacement, this demonstrates the likelihood of a peripheral resistance to thyroid hormones.
CURRENT BEST METHOD TO DIAGNOSIS
While a normal TSH cannot be used as a reliable indicator of global tissue thyroid effect, even a minimally elevated TSH (above 2 mU/L) demonstrates that there is a diminished intra-pituitary T3 level and is a clear indication (except in unique situations such as a TSH-secreting tumor) that the rest of the body is suffering from inadequate thyroid activity because the pituitary T3 level is always significantly higher than the rest of the body. Additionally, the most rigorously screened individuals for absence of thyroid disease have a TSH below 2 to 2.5 mU/L. Thus, treatment should be considered in any symptomatic person with a TSH greater than 2 mU/L. As discussed, a free T3/rT3 ratio below 0.2 is a useful indicator of low tissue thyroid levels. Additionally, a relatively low SHBG, a slow reflex relaxation time, a low resting metabolic rate (metabolism), and a low basal body temperature can also be useful indicators of low tissue thyroid levels and can aid in the diagnosis of tissue hypothyroidism.
CONCLUSION
The most important determinant of thyroid activity is the intra-cellular level of T3, and a major determinant of the intracellular T3 level is the activity of the cellular thyroid transporters. Reduced thyroid transport into the cell is seen with a wide range of common conditions, including insulin resistance, diabetes, depression, bipolar disorder, hyperlipidemia (high cholesterol and triglycerides), chronic fatigue syndrome, fibromyalgia, neurodegenerative diseases (Alzheimer’s, Parkinson’s and multiple sclerosis), migraines, stress, anxiety, chronic dieting and aging. The high incidence of reduced cellular thyroid transport seen with these conditions makes standard thyroid tests a poor indicator of cellular thyroid levels in these patient populations.
The pituitary has different transporters than every other tissue in the body; the thyroid transporters in the body are very energy dependent and are affected by numerous conditions while the pituitary is minimally affected. Because the pituitary remains largely unaffected, there is no elevation in TSH despite wide-spread tissue hypothyroidism. This explains why TSH is an inaccurate marker for tissue T3 levels for a variety of patients.
Reduced thyroid transport results in an artificial elevation in serum thyroid levels (especially T4), making this a poor marker for tissue thyroid levels, as well. Rather, an elevated or high-normal rT3 along with symptoms is shown to be a reliable marker for reduced transport of thyroid hormones and an indication that a person has low cellular thyroid levels despite having normal TSH, free T4, and free T3 levels (see Figure 2).
The intracellular T3 deficiency seen with these conditions often results in a vicious cycle of worsening symptoms that usually goes untreated because standard thyroid tests look normal. Additionally, it is not surprising that T4 preparations are generally ineffective in the presence of such conditions, while T3 replacement is shown to be beneficial, with potentially dramatic results. In the presence of such conditions, it should be understood that significant intracellular hypothyroidism may remain undiagnosed by standard blood tests. On the basis of the data presented here, the free T3/rT3 ratio and a low SHBG, along with signs and symptoms, including basal body temperature and the reflex relaxation phase, appear to be a more appropriate method for assessing the presence of hypothyroidism and determining whether supplementation with T3 (rather than T4 only) should be considered in a particular patient.
Thus, if a patient with a normal TSH presents with symptoms consistent with hypothyroidism, including fatigue, weight gain, depression, cold extremities, muscle aches, headaches, decreased libido, weakness, cold intolerance, water retention or PMS, a combination of both clinical and laboratory assessment should be used to determine the likely overall thyroid status and if a therapeutic trial of straight T3 or a T4/T3 combination is indicated.
DISCLOSURE OF INTERESTS
Dr. Holtorf supplies supplements from Holtraceuticals to support thyroid function to his own patients, outside the submitted work.
REFERENCES
- Everts ME, De Jong M, Lim CF, et al. Different regulation of thyroid hormone transport in liver and pituitary: its possible role in the maintenance of low T3 production during nonthyroidal illness and fasting in man.
- Peeters RP, Geyten SV, Wouters PJ, et al. Tissue thyroid hormone levels in critical illness.
- Lim C-F, Docter R, Krenning EP, et al. Transport of thyroxine into cultured hepatocytes: effects of mild nonthyroidal illness and calorie restriction in obese subjects.
- Sarne DH, Refetoff S. Measurement of thyroxine uptake from serum by cultured human hepatocytes as an index of thyroid status: reduced thyroxine uptake from serum of patients with nonthyroidal illness.
- Hennemann G, Docter R, Friesema EC, et al. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability.
- Holm AC, Jacquemin C. Membrane transport of L-triiodothyronine by human red cell ghosts.
- Docter R, Krenning EP, Bos G, Fekkes DSF, Hennemann G. Evidence that the uptake of triiodo-L-thyronine by human erythrocytes is carrier-mediated but not energy-dependent.
- Holm AC, Kagedal B. Kinetics of triiodothyronine uptake by erythrocytes in hyperthyroidism, hypothyroidism, and thyroid hormone resistance.
- Osty J, Valensi P, Samson M, Francon J, Blondeau JP. Transport of thyroid hormones by human erythrocytes: kinetic characterization in adults and newborns.
- Moreau X, Azorin J-M, Maurel M, Jeanningros R. Increase in red blood cell triiodothyronine uptake in untreated unipolar major depressed patients compared to healthy controls.
- Osty J, Jego L, Francon J, Blondeau JP. Characterization of triiodothyronine transport and accumulation in rat erythrocytes.
- Osty J, Zhou Y, Chantoux F, Francon J, Blondeau JP. The triiodothyronine carrier of rat erythrocytes: asymmetry and mechanism of transinhibition.
- Moreau X, Lejeune PJ, Jeanningros R. Kinetics of red blood cell T3 uptake in hypothyroidism with or without hormonal replacement, in the rat.
- McLeese JM, Eales JG. 3,5,3-Triiodo-L-thyronine and L-thyroxine uptake into red blood cells of rainbow trout (Oncorhynchus mykiss).
- McLeese JM, Eales JG. Characteristics of the uptake of 3,5,3-triiodo-L-thyronine and L-thyroxine into red blood cells of rainbow trout (Oncorhynchus mykiss).
- Everts ME, Docter R, van Buuren JC, et al. Evidence of carrier-mediated uptake of triiodothyronine in cultured anterior pituitary cells of euthyroid rats.
- Everts ME, Docter R, Moerings EP, van Koetsveld PM, Visser TJ, et al. Uptake of thyroxine in cultured anterior pituitary cells of euthyroid rats.
- Yan Z, Hinkle PM. Saturable, stereospecific transport of 3,5,3-triiodo-L-thyronine and L-thyroxine into GH4C1 pituitary cells.
- Goncalves E, Lakshmanan M, Pontecorvi A, Robbins J. Thyroid hormone transport in a human glioma cell line.
- Francon J, Chantoux F, Blondeau JP. Carrier-mediated transport of thyroid hormones into rat glial cells in primary culture.
- Beslin A, Chantoux F, Blondeau JP, Francon J. Relationship between the thyroid hormone transport system and the Na-H exchanger in cultured rat brain astrocytes.
- Chantoux F, Blondeau JP, Francon J. Characterization of the thyroid hormone transport system of cerebrocortical rat neurons in primary culture.
- Kastellakis A, Valcana T. Characterization of thyroid hormone transport in synaptosomes from rat brain.
- Lakshmanan M, Goncalves E, Lessly G, et al. The transport of thyroxine into mouse neuroblastoma cells, NB41A3: the effect of L-system amino acids.
- Pontecorvi A, Lakshmanan M, Robbins J. Intracellular transport of 3,5,3-triiodo-L-thyronine in rat skeletal myoblasts.
- Everts ME, Verhoeven FA, Bezstarosti K, et al. Uptake of thyroid hormones in neonatal rat cardiac myocytes.
- Zonefrati R, Rotella CM, Toccafondi RS, Arcangeli P. Thyroid hormone receptors in human cultured fibroblasts: evidence for cellular T4 transport and nuclear binding.
- Docter R, Krenning EP, Bernard HF, Hennemann G. Active transport of iodothyronines into human cultured fibroblasts.
- Cheng SY. Characterization of binding of uptake of 3,3,5-triiodo-L-thyronine in cultured mouse fibroblasts.
- Mitchell AM, Manley SW, Mortimer RH. Uptake of L-triiodothyronine by human cultured trophoblast cells.
- Mitchell AM, Manley SW, Mortimer RH. Membrane transport of thyroid hormone in the human choriocarcinoma cell line JAR.
- Mitchell AM, Manley SW, Rowan KA, Mortimer RH. Uptake of reverse T3 in the human choriocarcinoma cell line JAR.
- Bernus I, Mitchell AM, Manley SW, Mortimer RH. Uptake of L-triiodothyronine sulfate by human choriocarcinoma cell line JAR.
- Mitchell AM, Manley SW, Payne EJ, Mortimer RH. Uptake of thyroxine in the human choriocarcinoma cell line JAR.
- Landeta LC, Gonzales-Padrones T, Rodriguez-Fernandez C. Uptake of thyroid hormones (L-T3 and L-T4) by isolated rat adipocytes.
- Kostrouch Z, Felt V, Raska J, Nedvidkova J, Holeckova E. Binding of (125I) triiodothyronine to human peripheral leukocytes and its internalization.
- Kostrouch Z, Raka I, Felt V, Nedvidkova J, Holeckova E. Internalization of triiodothyronine-bovine serum albumin-colloidal gold complexes in human peripheral leukocytes.
- Centanni M, Mancini G, Andreoli M. Carrier-mediated [125I]-T3 uptake by mouse thymocytes.
- Centanni M, Sapone A, Taglienti A, Andreoli M. Effect of extracellular sodium on thyroid hormone uptake by mouse thymocytes.
- de Jong M, Docter R, Bernard HF, et al. T4 uptake into the perfused rat liver and liver T4 uptake in humans are inhibited by fructose.
- Hennenmann G, Everts ME, de Jong M, et al. The significance of plasma membrane transport in the bioavailability of thyroid hormone.
- Vos RA, de Jong M, Bernard BF, et al. Impaired thyroxine and 3,5,3′-triiodothyronine handling by rat hepatocytes in the presence of serum of patients with nonthyroidal illness.
- Hennemann G, Krenning EP. The kinetics of thyroid hormone transporters and their role in non-thyroidal illness and starvation.
- Hennemann G, Vos RA, de Jong M, et al. Decreased peripheral 3,5,3′-triiodothyronine (T3) production from thyroxine (T4): a syndrome of impaired thyroid hormone activation due to transport inhibition of T4- into T3-producing tissues.
- Stump CS, Short KR, Bigelow ML, et al. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts.
- Krenning EP, Docter R, Bernard HF, et al. The essential role of albumin in the active transport of thyroid hormones into primary cultured rat hepatocytes.
- Krenning EP, Docter R, Bernard HF, et al. Regulation of the active transport of 3,3′,5-triiodothyronine (T3) into primary cultured rat hepatocytes by ATP.
- van der Heyden JT, Docter R, van Toor H, et al. Effects of caloric deprivation on thyroid hormone tissue uptake and generation of low-T3 syndrome.
- Wassen FWJS, Moerings EPCM, van Toor H, et al. Thyroid hormone uptake in cultured rat anterior pituitary cells: effects of energy status and bilirubin.
- Jenning AS, Ferguson DC, Utiger RD. Regulation of the conversion of thyroxine to triiodothyronine in the perfused rat liver.
- Krenning E, Docter R, Bernard B, Visser T, Hennemann G. Characteristics of active transport of thyroid hormone into rat hepatocytes.
- Riley WW, Eales JG. Characterization of 3,5,3-triiodo-L-thyronine transport into hepatocytes isolated from juvenile rainbow trout (Oncorhynchus mykiss), and comparison with L-thyroxine transport.
- Spencer CA, Lum SMC, Wilber JF, et al. Dynamics of serum thyrotropin and thyroid hormone changes in fasting.
- St Germain DL, Galton VA. Comparative study of pituitary-thyroid hormone economy in fasting and hypothyroid rats.
- Arem R, Wiener GJ, Kaplan SG, Kim HS, et al. Reduced tissue thyroid hormone levels in fatal illness.
- Lim C-F, Bernard BF, De Jong M, et al. A furan fatty acid and indoxyl sulfate are the putative inhibitors of thyroxine hepatocyte transport in uremia.
- Lim C-F, Docter R, Visser TJ, Krenning EP, Bernard B, et al. Inhibition of thyroxine transport into cultured rat hepatocytes by serum of non-uremic critically ill patients: effects of bilirubin and nonesterified fatty acids.
- Lim VS, Passo C, Murata Y, Ferrari E, et al. Reduced triiodothyronine content in liver but not pituitary of the uremic rat model: demonstration of changes compatible with thyroid hormone deficiency in liver only.
- Everts ME, Lim C-F, Moerings EPCM, Docter R, et al. Effects of a furan fatty acid and indoxyl sulfate on thyroid hormone uptake in cultured anterior pituitary cells.
- Doyle D. Benzodiazepines inhibit temperature dependent L-[125I] triiodothyronine accumulation into human liver, human neuroblast, and rat pituitary cell lines.
- Krenning EP, Docter R, Bernard HF, et al. Decreased transport of thyroxine (T4), 3,3′,5-triiodothyronine (T3) and 3,3′,5′-triiodothyronine (rT3) into rat hepatocytes in primary culture due to a decrease of cellular ATP content and various drugs.
- Kaptein EM, Robinson WJ, et al. Peripheral serum thyroxine, triiodothyronine, and reverse triiodothyronine in the low thyroxine state of acute nonthyroidal illness. A noncompartmental analysis.
- Kaptein EM, Kaptein JS, Chang EI, et al. Thyroxine transfer and distribution in critical nonthyroidal illness, chronic renal failure, and chronic ethanol abuse.
- Everts ME, Visser TJ, Moerings EM, Docter R, et al. Uptake of triiodothyroacetic acid and its effect on thyrotropin secretion in cultered anterior pituitary cells.
- De Jong M, Docter R, van der Hoek HJ, Vos RA. Transport of 3,5,3′-triiodothyronine into the perfused rat liver and subsequent metabolism are inhibited by fasting.
- Hennemann G, Krenning EP, Bernard B, Huvers F, et al. Regulation of Influx and efflux of thyroid hormones in rat hepatocytes: possible physiologic significance of plasma membrane in the regulation of thyroid hormone activity.
- Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of Type 2 diabetic parents.
- Szendroedi J, Schmid AI, Meyerspeer M, et al. Impaired mitochondrial function and insulin resistance of skeletal muscle in mitochondrial diabetes.
- Abdul-Ghani MA, Jani R, Chavez A, et al. Mitochondrial reactive oxygen species generation in obese non-diabetic and type 2 diabetic participants.
- Verga SB, Donatelli M, Orio L, et al. A low reported energy intake is associated with metabolic syndrome.
- Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease.
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine.
- Fosslien E. Mitochondrial medicine – Molecular pathology of defective oxidative phosphorylation.
- West IC. Radicals and oxidative stress in diabetes.
- Brehm A, Krssak M, Schmid AI, et al. increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle.
- DeMarco NM, Beitz DC, Whitehurst GB. Effect of fasting on free fatty acid, glycerol and cholesterol concentrations in blood plasma and lipoprotein lipase activity in adipose tissue of cattle.
- Leibel RL, Jirsch J. Diminshed energy requirements in reduced-obese patients.
- Steen SN, Opplieger RA, Brownell KD. Metabolic effects of repeated weight and regain in adolescent wrestlers.
- Elliot DL, Goldberg L, Kuehl KD, Bennett WM. Sustained depression of the resting metabolic rate after massive weight loss.
- Manore MM, Berry TE, Skinner JS, Carroll SS. Energy expenditure at rest and during exercise in nonobese female cyclical dieters and in nondieting control subjects.
- Croxson MS, Ibbertson HK. Low serum triiodothyronine (T3) and hypothyroidism in anorexia nervosa.
- Carlin K, Carlin S. Possible etiology for euthyroid sick syndrome.
- Brownell KD, Greenwood MR, Stellar E, Shrager EE. The effects of repeated cycles of weight loss and regain in rats.
- Modica-Napolitano JS, Renshaw PF. Ethanolamine and phosphoethanolamine inhibit mitochondrial function in vitro: implications for mitochondrial dysfunction hypothesis in depression and bipolar disorder.
- Gardner A, Boles RG. Mitochondrial energy depletion in depression with somatization.
- Burroughs S, French D. Depression and anxiety: role of mitochondria.
- Einat H, Yuan P, Manji HK. Increased anxiety-like behaviors and mitochondrial dysfunction in mice with targeted mutation of the Bcl-2 gene: further support for the involvement of mitochondrial function in anxiety disorders.
- Stork C, Renshaw PF. Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research.
- Fattal O, Budur K, Vaughan AJ, Franco K. Review of the literature on major mental disorders in adult patients with mitochondrial diseases.
- Hutchin T, Cortopassi G. A mitochondrial DNA clone is associated with increased risk for Alzheimer’s disease.
- Sherer TB, Betarbet R, Greenamyre JT. Environment, mitochondria, and Parkinson’s disease.
- Gomez C, Bandez MJ, Navarro A. Pesticides and impairment of mitochondrial function in relation with the Parkinsonian syndrome.
- Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death?
- Schapira AHV. Mitochondrial disease.
- Richter C. Oxidative damage to mitochondrial DNA and its relationship to aging.
- Papa S. Mitochondrial oxidative phosphorylation changes in the life span. Molecular aspects and physiopathological implications.
- Cortopassi G, Wang A. Mitochondria in organismal aging and degeneration.
- Harman D. The biologic clock: the mitochondria?
- Miquel J, Economos AC, Fleming J, Johnson JE. Mitochondrial role in cell aging.
- Miquel J. An integrated theory of aging as the result of mitochondrial DNA mutation in differentiated cells.
- Miquel J. An update on the mitochondrial-DNA mutation hypothesis of cell aging.
- Zs.-Nagy I. A membrane hypothesis of aging.
- Zs.-Nagy I. The role of membrane structure and function in cellular aging: a review.
- Savitha S, Sivarajan K, Haripriya D, et al. Efficacy of levo carnitine and alpha lipoic acid in ameliorating the decline in mitochondrial enzymes during aging.
- Skulachev VP, Longo VD. Aging as a mitochondria-mediated atavistic program: can aging be switched off?
- Corral-Debrinski M, Shoffner JM, Lott MT, Wallace DC. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease.
- Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging.
- Fulle S, Mecocci P, Fano G, et al. Specific oxidative alterations in vastus lateralis muscle of patients with the diagnosis of chronic fatigue syndrome.
- Buist R. Elevated xenobiotics, lactate and pyruvate in C.F.S. patients.
- Park JH, Niermann KJ, Olsen N. Evidence for metabolic abnormalities in the muscles of patients with fibromyalgia.
- Yunus MB, Kalyan-Raman UP, Kalyan-Raman K. Primary fibromyalgia syndrome and myofascial pain syndrome: clinical features and muscle pathology.
- Puddu P, Puddu GM, Galletti L, Cravero E, Muscari A. Mitochondrial dysfunction as an initiating event in atherogenesis: a plausible hypothesis.
- Chen L, Knowlton AA. Depressed mitochondrial fusion in heart failure.
- Kaptein EM, Feinstein EI, Nicoloff JT, Massry SG. Serum reverse triiodothyronine and thyroxine kinetics in patients with chronic renal failure.
- Kaptein EM. Thyroid hormone metabolism and thyroid disease in chronic renal failure.
- Kaptein EM. Clinical relevance of thyroid hormone alterations in nonthyroidal illness.
- Kigoshi S, Akiyama M, Ito R. Close correlation between levels of cholesterol and free fatty acids in lymphoid cells.
- Bochukova E, Schoenmakers N, Agostini M, et al. A mutation in the thyroid hormone receptor alpha gene.
- Meier C, Trittibach P, Guglielmetti M, Staub JJ, Muller B. Serum TSH in assessment of severity of tissue hypothyroidism in patients with overt primary thyroid failure: cross sectional survey.
- Alevizaki M, Mantzou E, cimponeriu AT, et al. TSH may not be a good marker for adequate thyroid hormone replacement therapy.
- Zulewski H, Muller B, Exer P, et al. Estimation of tissue hypothyrodisim by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls.
- van den Beld AW, Visser TJ, Feelders RA, et al. Thyroid hormone concentrations, disease, physical function and mortality in elderly men.
- Singer PA, Cooper DS, Levy EG, et al. for the Standards of Care Committee, American Thyroid Association. Treatment guidelines for patients with hyperthyroidism and hypothyroidism.
- Escobar-Morreale HF, Obregón MJ, Escobar del Rey F, et al. Replacement therapy for hypothyroidism with thyroxine alone does not ensure euthyroidism in all tissues, as studied in thyroidectomized rats.
- Escobar-Morreale HF, Obregón MJ, Escobar del Rey F. Only the combined treatment with thyroxine and triiodothyronine ensures euthyroidism in all tissues of the thyroidectomized rat.
- Fraser WD, Biggart EM, OReilly DJ, et al. Are biochemical tests of thyroid function of any value in monitoring patients receiving thyroxine replacement?
- Lecomte P, Lecureuil N, Lecureuil M, Salazar CO, Valat C. Age modulates effects of thyroid dysfunction on sex hormone binding globulin (SHBG) levels.
- Sarne DH, Refetoff S, Rosenfield RL, Farriaux JP. Sex hormone-binding globulin in the diagnosis of peripheral tissue resistance to thyroid hormone: the valued of changes after short term triiodothyronine administration.
- Krotkiewski M, Holm G, Shono N. Small doses of triiodothyronine can change some risk factors associated with abdominal obesity.
- Birkeland KI, Hanssen KF, Torjesen PA, et al. Level of Sex Hormone-binding globulin is positively correlated with insulin sensitivity in men with Type 2 diabetes.
- Chen BH, Brennan K, Goto A. Sex hormone-binding globulin and risk of clinical diabetes in American black, Hispanic, and Asian/Pacific Islander postmenopausal women.
- Moncay H, Dapunt O, Moncayo R. diagnostic accuracy of basal TSH determinations based on the intravenous TRH stimulation tests: an evaluation of 2570 tests and comparison with the literature.
- Zulewski H, Müller B, Exer P, Miserez AR, Staub JJ. Estimation of tissue hypothyroidism by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls.
- Al-Adsani H, Hoffer LJ, Silva JE. Resting energy expenditure is sensitive to small dose changes in patients on chronic thyroid hormone replacement.
- Barnes B. Temperature versus basal metabolism.
- Friedman M, Miranda-Massari JR, Gonzalez MJ. Supraphysiological cyclic dosing of sustained release T3 in order to reset low basal body temperature.
- Hackney AC, Feith S, Pozos R, Seale J. Effects of high altitude and cold exposure on resting thyroid hormone concentrations.
- Opstad PK, Falch D, Oktedalen O, et al. The thyroid function in young men during prolonged exercise and the effect of energy and sleep deprivation.
- Ellingsen DG, Efskind J, Haug E, et al. Effects of low mercury vapour exposure on the thyroid fucniton in Chloralkai workers.
- den Brinker M, Joosten KFM, Visser TJ, et al. Euthyroid sick syndrome in meningococcal sepsis: the impact of peripheral thyroid hormone metabolism and binding proteins.
- Chopra IJ, Solomon DH, Hepner GW, et al. Misleadingly low free thyroxine index and usefulness of reverse triiodothyronine measurement in nonthyroidal illnesses.
- Chopra IJ. A study of extrathyroidal conversion of thyroxine (T4) to 3,3′,5-triiodothyronine (T3) in vitro.
- Sechman A, Niezgoda J, Sobocinski R. The relationship between basal metabolic rate (BMR) and concentrations of plasma thyroid hormones in fasting cockerels.
- Magri F, Cravello L, Fioravanti M, et al. Thyroid function in old and very old healthy subjects.
- O’Brian JI, Baybee DE, Wartofsky L, et al. Altered peripheral thyroid hormone metabolism and diminished hypothalamic pituitary responsiveness with changes in dietary composition.
- Friberg L, Drvota V, Bjelak AH, Eggertsen G, Ahnve S. Association between increased levels of reverse triiodothyronine and mortality after acute myocardial infarction.
- McCormack PD. Cold stress, reverse T3 and lymphocyte function.
- Scriba PC, Bauer M, Emmert D, et al. Effects of obesity, total fasting and re-alimentation on L-thyroxine (T4), 3,5,3-L-triiodothyronine (T3), 3,3,5-L-triiodothyronine (rT3), thyroxine binding globulin (TBG), transferrin, 2-haptoglobin and complement C3 in serum.
- Kvetny J. Thyroxine binding and cellular metabolism of thyroxine in mononuclear blood cells from patients with anorexia nervosa.
- Germain DL. Metabolic effect of 3,3′,5′-triiodothyronine in cultured growth hormone-producing rat pituitary tumor cells. Evidence for a unique mechanism of thyroid hormone action.
- Szymanski PT, Nauman J. Effects of thyroid hormones and reverse T3 pretreatment on the beta adrenoreceptors in the rat heart.
- du Pont JS. Is reverse T3 a physiological nonactive competitor of the action of T3 upon the electrical properties of GH3 cells?
- Schulte C. Low T3 syndrome and nutritional status as prognostic factors in patients undergoing bone marrow transplantation.
- Goichot B, Schlienger JL, Grunenberger F, et al. Thyroid hormone status and nutrient intake in the free-living elderly. Interest of reverse triiodothyronine assessment.
- Okamoto R, Leibfritz D. Adverse effects of reverse triodothyronine on cellular metabolism as assessed by 1H and 31P NMR spectroscopy.
- de Jong FJ, den Heijer T, Visser TJ, et al. Thyroid hormones, dementia, and atrophy of the medial temporal lobe.
- Forestier E, Vinzio S, Sapin R, et al. Increased reverse T3 is associated with shorter survival in independently-living elderly. The Alsanut Study.
- Visser TJ, Lamberts WJ, Wilson JHP, et al. Serum thyroid hormone concentrations during prolonged reduction of dietary intake.
- Linnoila M, Lamberg BA, Potter WZ, et al. High reverse T3 levels in manic and unipolar depressed women.
- McCormack PD, Reed HL, Thomas JR, et al. Increased in rT3 serum levels observed during extended Alaskan field operations of naval personnel.
- Mariotti S, Barbesino G, Caturegli P, et al. Complex alteration of thyroid function in healthy centenarians.
- Danforth EJ, Desilets EJ, Jorton ES, et al. Reciprocal serum triiodothryronine (T3) and reverse (rT3) induced by altering the carbohydrate content of the diet.
- McCormack PD, Thomas J, Malik M, Staschen CM. Cold stress, reverse T3 and lymphocyte function.
- Peeters RP, Wouters PJ, van Toor H, et al. Serum 3,3′,5′-triiodothyronine (rT3) and 3,5,3′-triiodothyronine/rT3 are prognostic markers in critically ill patients and are associated with postmortem tissue deiodinase activities.
- Szabolcs I, Weber M, Kovacs Z, et al. The possible reason for serum 3,3′5′-(reverse T3) triiodothyronine increase in old people.
- Silberman H, Eisenberg D, Ryan J, et al. The relation of thyroid indices in the critically ill patient to prognosis and nutritional factors.
- Stan M, Morris JC. Thyrotropin-axis adaptation in aging and chronic disease.
- LoPresti JS, Eigen A, Kaptein E, et al. Alterations in 3,3′,5′-Triiodothyronine metabolism in response to propylthiouracil, dexamethasone, and thyroxine administration in man.
- Palmblad J, Levi L, Burger A, et al. Effects of total energy withdrawal (fasting) on the levels of growth hormone, thyrotropin, cortisol, adrenaline, noradrenaline, T4, T3, and rT3 in healthy males.
- Reinhardt W, Misch C, Jockenhovel F, et al. Triiodothyronine (T3) reflects renal graft function after renal transplantation.
- Chopra IJ, Chopra U, Smith SR, et al. Reciprocal changes in serum concentrations of 3,3′5′-triiodothyronine (reverse T3) and 3,3′5-triiodothyronine (T3) in systemic illnesses.
- Spaulding SW, Chopra IJ, Swherwin RS, et al. Effect of caloric restriction and dietary compostion on serum T3 and reverse T3 in man.
- Girdler SS, Pedersen CA, Light KC. Thyroid axis function during the menstrual cycle in women with premenstrual syndrome.
- Peeters RP, Wouters PJ, Kaptein E, et al. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients.
- Pittman JA, Tingley JO, Nickerson JF, Hill SR. Antimetabolic activity of 3,3′,5′-triiodo-DL-thyronine in man.
- Desai M, Irani AJ, Patil K, et al. The importance of reverse triiodothyroinine in hypothyroid children on replacement treatement.
- Chopra IJ. A radioimmunoassay for measurement of 3,3′,5′-triiodothyronine (reverse T3).
- Kodding R, Hesch RD. L-3′,5′-diiodothyronine in human serum.
- Benua RS, Kumaoka S, Leeper RD, Rawson RW. The effect of DL-3,3′,5′-triiodothyronine in Grave’s disease.
- Chopra IJ. Study of extrathyroidal conversion of T4 to T3 in vitro: evidence that reverse T3 is a potent inhibitor of T3 production.
- Gavin LA, Moeller M, Shoback D, Cavalieri RR. Reverse T3 and modulators of the calcium messenger system rapidly decrease T4-5′-deiodinase II activity in cultured mouse neuroblastoma cells.
- Chopra IJ, Williams DE, Orgiazzi J, Solomon DH. Opposite effects of dexamethasone on serum concentrations of 3,3′,5′-triiodothyronine (reverse T3) and 3,3′5-triiodothyronine (T3).
- Brent GA, Hershman JM. Thyroxine therapy in patients with severe nonthyroidal illnesses and low serum thyroxine concentration.
- Lomenick JP, El-Sayyid M, Smith WJ. Effect of levo-thyroxine treatment on weight and body mass index in children with acquired hypothyroidism.
- Acker CG, Singh AR, Flick RP, et al. A trial of thyroxine in acute renal failure.
- Samuels MH, Schuff KG, Carlson NE, Carello P, Janowsky JS. Health status, psychological symptoms, mood, and cognition in L-thyroxine-treated hypothyroid subjects.
- Krotkiewski M. Thyroid hormones and treatment of obesity.
- Lowe JC, Garrison RL, Reichman AJ, et al. Effectiveness and safety of T3 (triiodothyronine) therapy for euthyroid fibromyalgia: a double-blind placebo-controlled response-driven crossover study.
- Lowe JC, Reichman AJ, Yellin J. The process of change during T3 treatment for euthyroid fibromyalgia: a double-blind placebo-controlled crossover study.
- Lowe JC, Garrison RL, Reichman AJ, et al. Triiodothyronine (T3) treatment of euthyroid fibromyalgia: a small-n replication of a double-blind placebo-controlled crossover study.
- Yellin BA, Reichman AJ, Lowe JC. The process of Change During T3 treatment for euthyroid fibromyalgia: a double-blind placebo-controlled crossover study.
- Cooke RG, Joffe RT, Levitt AJ. T3 augmentation of antidepressant treatment in T4-replaced thyroid patients.
- Bettendorf M, Schmidt KG, Grulich-Henn J, et al. Tri-idothyronine treatment in children after cardiac surgery: a double-blind, randomized, placebo-controlled study.
- Pingitore A, Galli E, Barison A, et al. Acute effects of triiodothryronine replacement therapy in patients with chronic heart failure and low-T3 syndrome: a randomized placebo-controlled study.
- Meyer T, Husch M, van den Berg E, et al. Treatment of dopamine-dependent shock with triiodothyronine: preliminary results.
- Dulchavsky SA, Hendrick SR, Dutta S. Pulmonary biophysical effects of triiodothyronine (T3) augmentation during sepsis induced hypothyroidism.
- Novitzsky D, Cooper DKC, Human PA, et al. Triiodothyronine therapy for heart donor and recepient.
- Dulchavsky SA, Maitra SR, Maurer J, et al. Beneficial effects of thyroid hormone administration in metabolic and hemodynamic function in hemorrhagic shock.
- Klemperer JD, Klein I, Gomez M, et al. Thyroid hormone treatment after coronary-artery bypass surgery.
- Gomberg-Maitland M. Thyroid hormone and cardiovascular disease.
- Dulchavsky SA, Kennedy PR, Geller ER, et al. T3 preserves respiratory function in sepsis.
- Novitzky D, Cooper DK, Reichart B. Hemodynamic and metabolic responses to hormonal therapy in brain-dead potential organ donors.
- Hamilton MA, Stevenson LW, Fonarow GC, et al. Safety and hemodynamic effects of intravenous triiodothyronine in advanced congestive heart failure.
- Klemperer JD, Klein IL, Ojamaa K, et al. Triidothyronine therapy lowers the incidence of atrial fibrillation after cardiac operations.